

Glucocorticoid-induced autophagy in osteocytes
- PMID: 20564240
- PMCID: PMC3179284
- DOI: 10.1002/jbmr.160
Glucocorticoid-induced autophagy in osteocytes
Abstract
Glucocorticoid (GC) therapy is the most frequent cause of secondary osteoporosis. In this study we have demonstrated that GC treatment induced the development of autophagy, preserving osteocyte viability. GC treatment resulted in an increase in autophagy markers and the accumulation of autophagosome vacuoles in vitro and in vivo promoted the onset of the osteocyte autophagy, as determined by expression of autophagy markers in an animal model of GC-induced osteoporosis. An autophagy inhibitor reversed the protective effects of GCs. The effects of GCs on osteocytes were in contrast to tumor necrosis factor α (TNF-α), which induced apoptosis but not autophagy. Together this study reveals a novel mechanism for the effect of GC on osteocytes, shedding new insight into mechanisms responsible for bone loss in patients receiving GC therapy.
© 2010 American Society for Bone and Mineral Research.
Figures
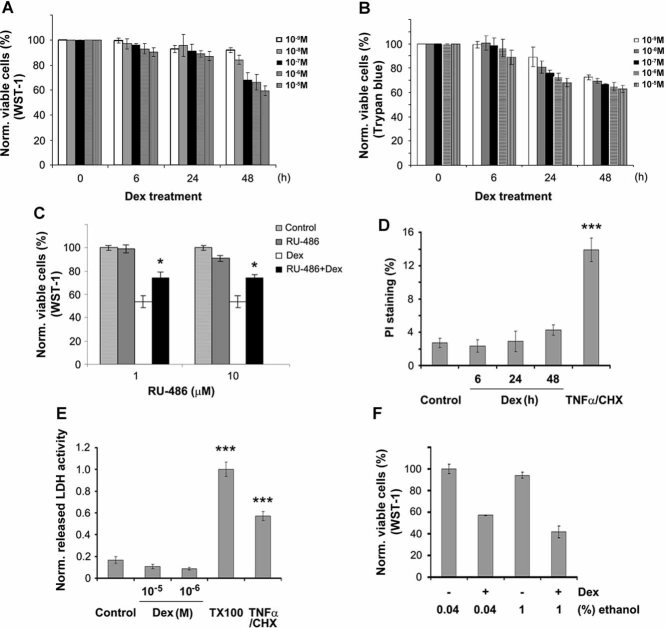
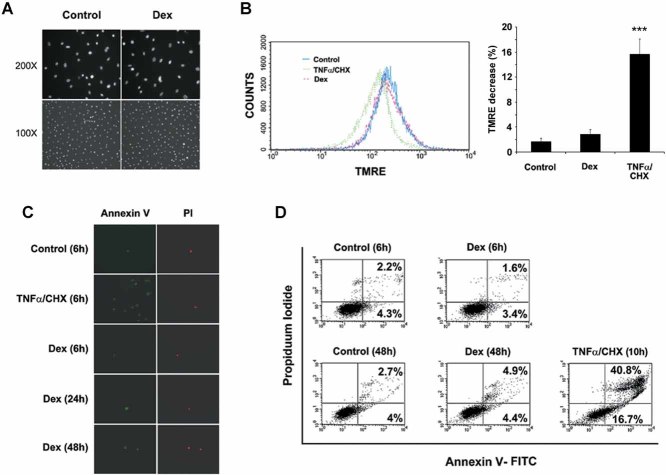
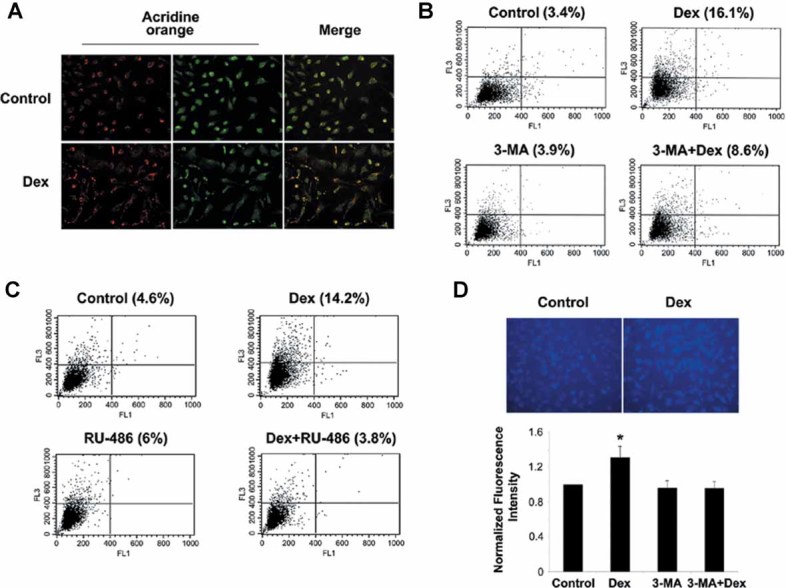
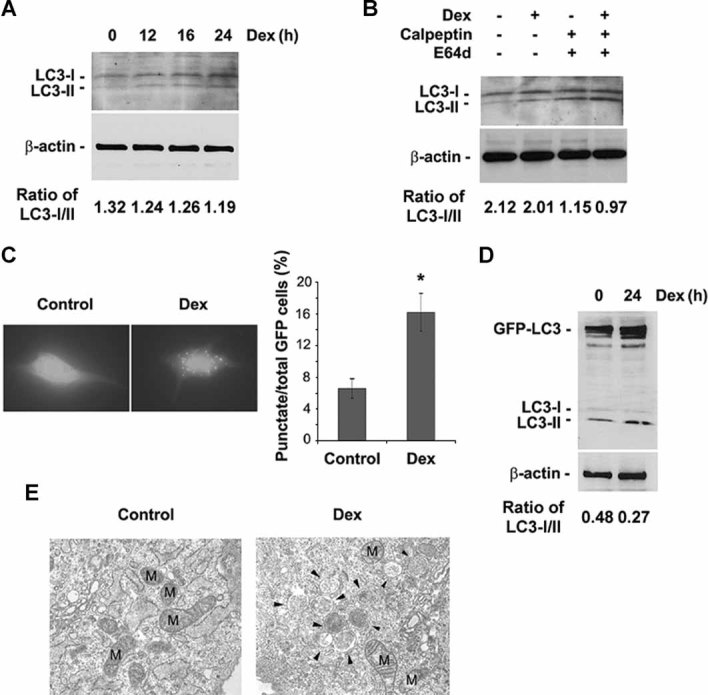
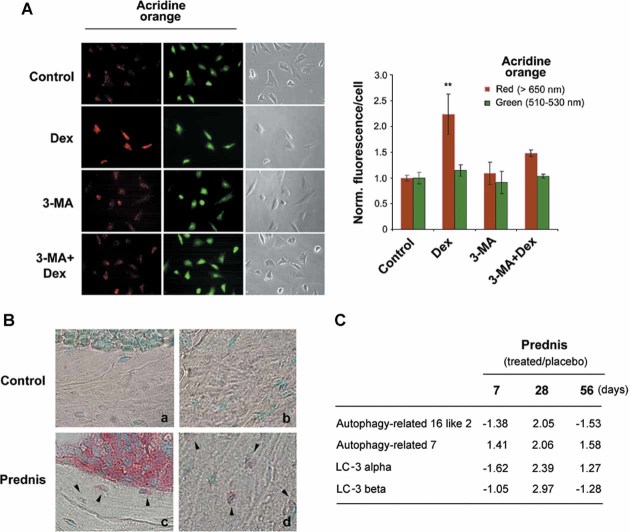
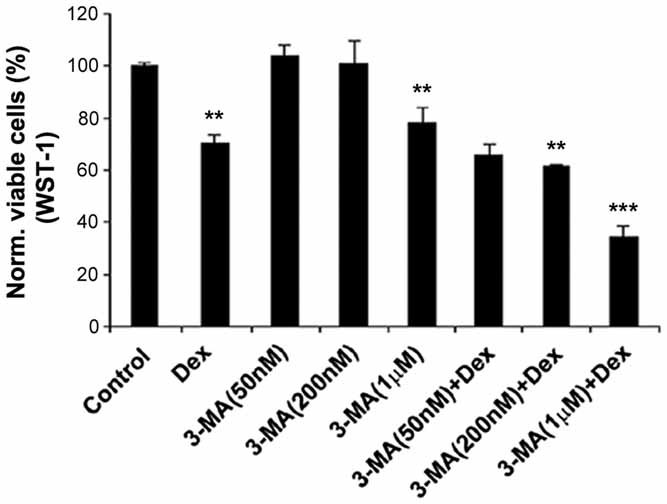
References
-
- Weinstein R. Glucocorticoid-induced osteoporosis. In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Washington, DC: American Society of Bone and Mineral Research; 2008.
-
- Canalis E, Delany AM. Mechanisms of glucocorticoid action in bone. Ann NY Acad Sci. 2002;966:73–81. - PubMed
-
- O'Brien CA, Jia D, Plotkin LI, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–1841. - PubMed
-
- Gohel A, McCarthy MB, Gronowicz G. Estrogen prevents glucocorticoid-induced apopotosis in osteoblasts in vivo and in vitro. Endocrinology. 1999;140:5339–5347. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
