

Regulation of cardiac gene expression by KLF15, a repressor of myocardin activity
- PMID: 20566642
- PMCID: PMC2930743
- DOI: 10.1074/jbc.M110.107292
Regulation of cardiac gene expression by KLF15, a repressor of myocardin activity
Abstract
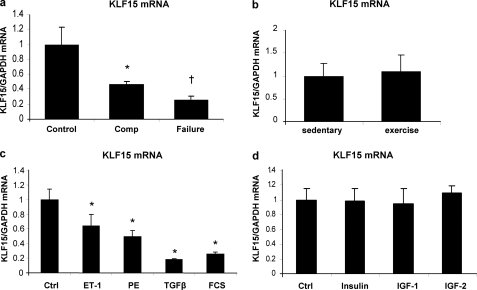
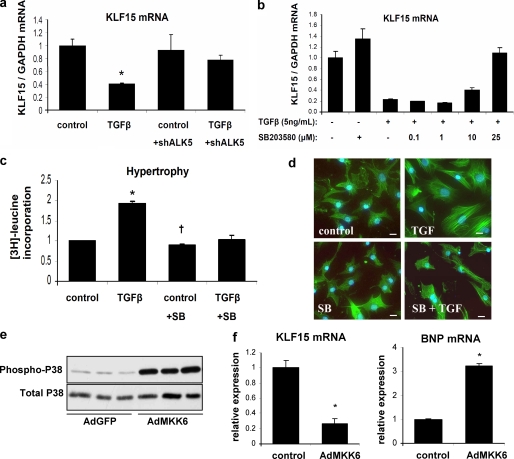
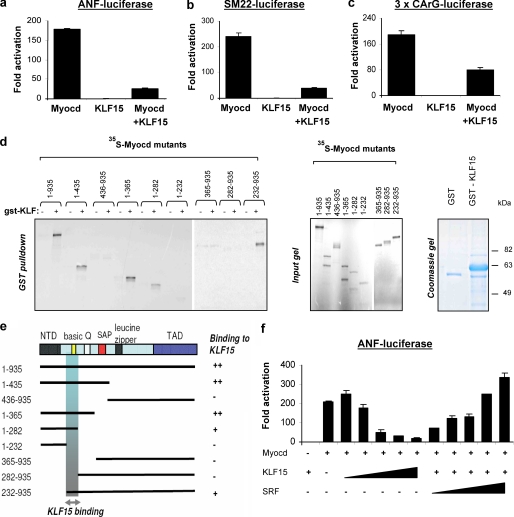
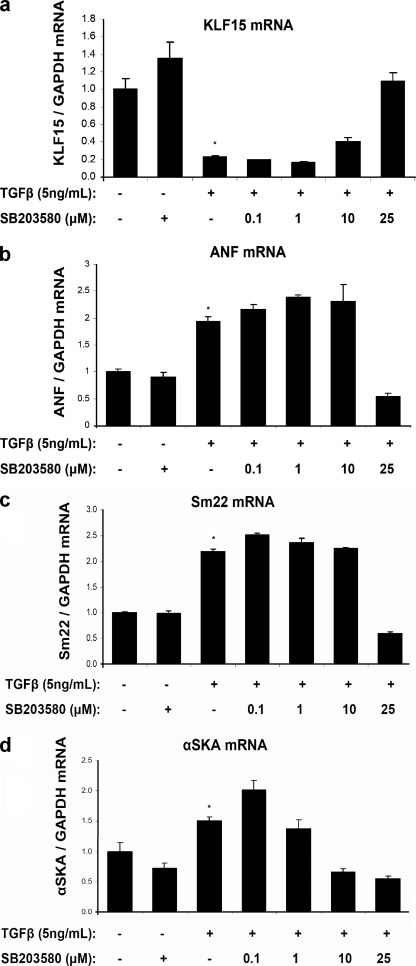
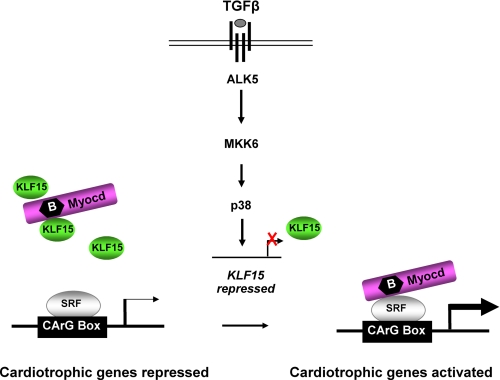
Pathological forms of left ventricular hypertrophy (LVH) often progress to heart failure. Specific transcription factors have been identified that activate the gene program to induce pathological forms of LVH. It is likely that apart from activating transcriptional inducers of LVH, constitutive transcriptional repressors need to be removed during the development of cardiac hypertrophy. Here, we report that the constitutive presence of Krüppel-like factor 15 (KLF15) is lost in pathological hypertrophy and that this loss precedes progression toward heart failure. We show that transforming growth factor-beta-mediated activation of p38 MAPK is necessary and sufficient to decrease KLF15 expression. We further show that KLF15 robustly inhibits myocardin, a potent transcriptional activator. Loss of KLF15 during pathological LVH relieves the inhibitory effects on myocardin and stimulates the expression of serum response factor target genes, such as atrial natriuretic factor. This uncovers a novel mechanism where activated p38 MAPK decreases KLF15, an important constitutive transcriptional repressor whose removal seems a vital step to allow the induction of pathological LVH.
Figures
References
-
- Oka T., Maillet M., Watt A. J., Schwartz R. J., Aronow B. J., Duncan S. A., Molkentin J. D. (2006) Circ. Res. 98, 837–845 - PubMed
-
- Zhang X., Azhar G., Chai J., Sheridan P., Nagano K., Brown T., Yang J., Khrapko K., Borras A. M., Lawitts J., Misra R. P., Wei J. Y. (2001) Am. J. Physiol. Heart Circ. Physiol. 280, H1782–H1792 - PubMed
-
- Wang D., Chang P. S., Wang Z., Sutherland L., Richardson J. A., Small E., Krieg P. A., Olson E. N. (2001) Cell 105, 851–862 - PubMed
-
- Pipes G. C., Creemers E. E., Olson E. N. (2006) Genes Dev. 20, 1545–1556 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
