

Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease
- PMID: 20566712
- PMCID: PMC2916709
- DOI: 10.1093/hmg/ddq253
Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease
Abstract
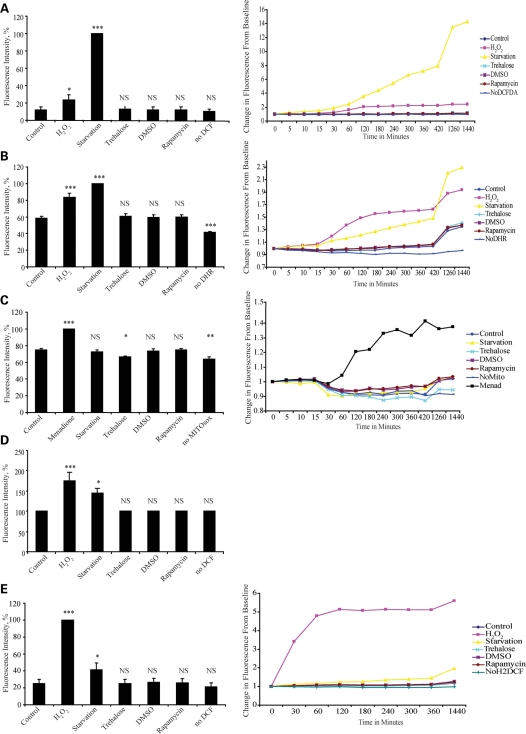
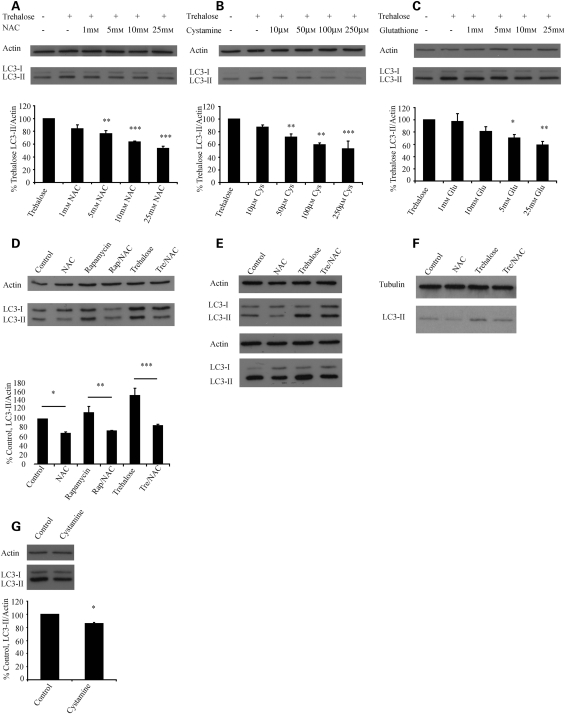
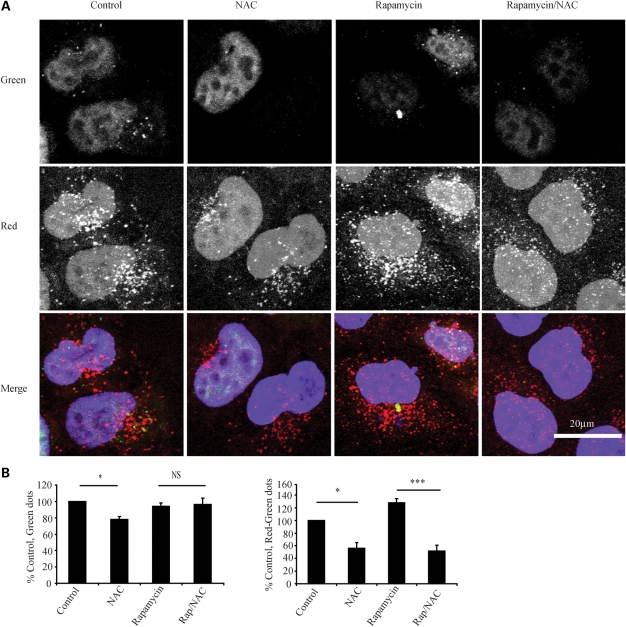
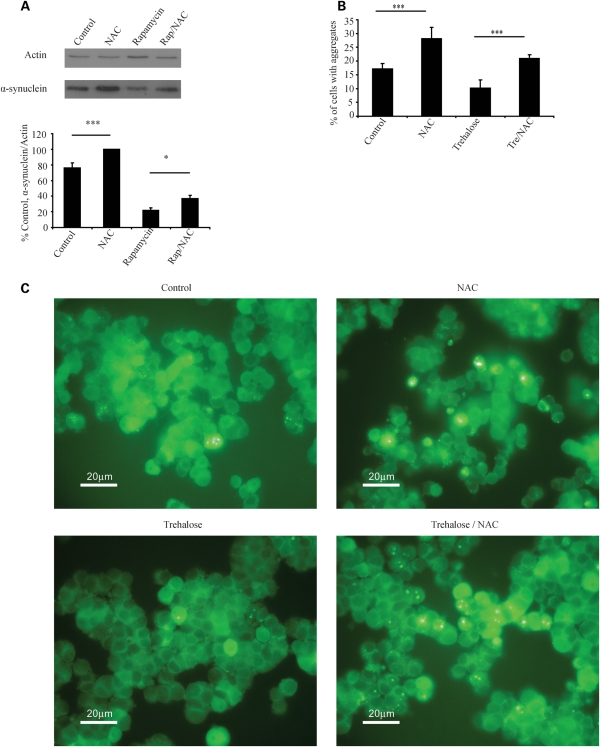
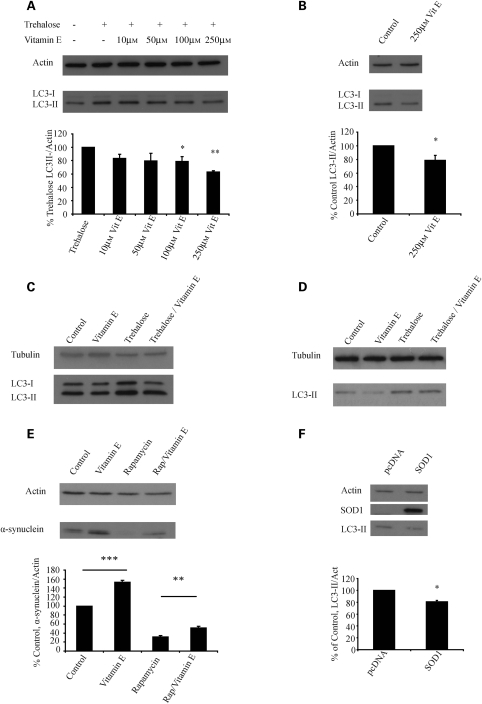
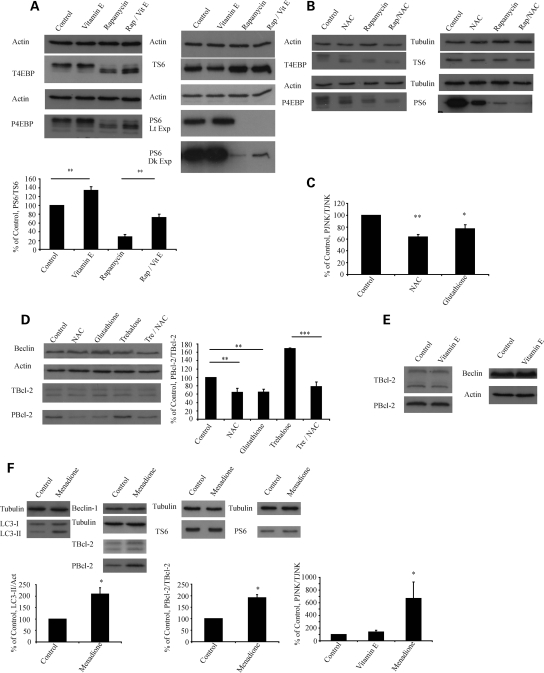
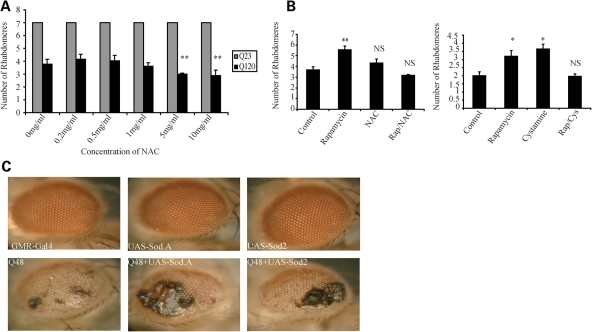
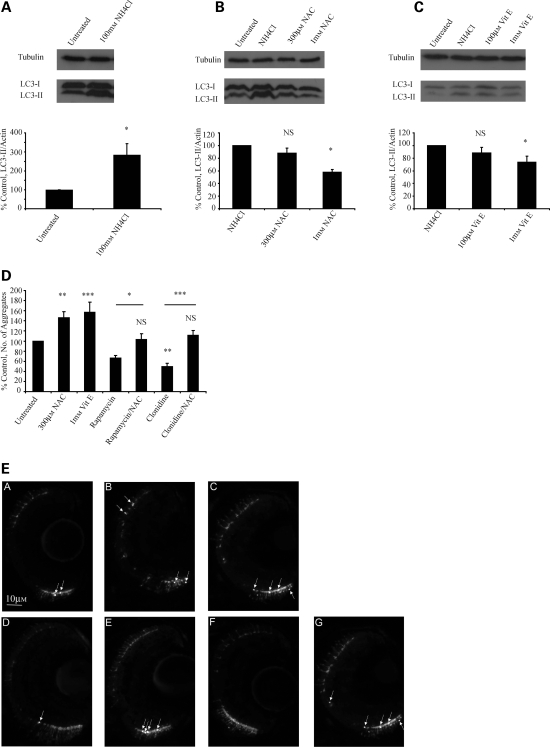
Many neurodegenerative diseases exhibit protein accumulation and increased oxidative stress. Therapeutic strategies include clearing aggregate-prone proteins by enhancing autophagy or decreasing oxidative stress with antioxidants. Many autophagy-inducing stimuli increase reactive oxygen species (ROS), raising concerns that the benefits of autophagy up-regulation may be counterbalanced by ROS toxicity. Here we show that not all autophagy inducers significantly increase ROS. However, many antioxidants inhibit both basal and induced autophagy. By blocking autophagy, antioxidant drugs can increase the levels of aggregate-prone proteins associated with neurodegenerative disease. In fly and zebrafish models of Huntington's disease, antioxidants exacerbate the disease phenotype and abrogate the rescue seen with autophagy-inducing agents. Thus, the potential benefits in neurodegenerative diseases of some classes of antioxidants may be compromised by their autophagy-blocking properties.
Figures
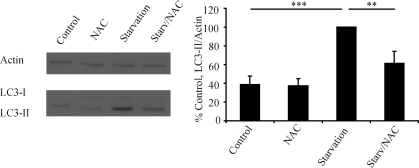
Similar articles
-
Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies.Cell Death Differ. 2009 Jan;16(1):46-56. doi: 10.1038/cdd.2008.110. Epub 2008 Jul 18. Cell Death Differ. 2009. PMID: 18636076 Review.
-
Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease.Redox Biol. 2013 Dec 25;2:82-90. doi: 10.1016/j.redox.2013.12.013. eCollection 2014. Redox Biol. 2013. PMID: 24494187 Free PMC article. Review.
-
Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia.J Neurol Sci. 2005 Jun 15;233(1-2):145-62. doi: 10.1016/j.jns.2005.03.012. J Neurol Sci. 2005. PMID: 15896810 Review.
-
AUTEN-67 (Autophagy Enhancer-67) Hampers the Progression of Neurodegenerative Symptoms in a Drosophila model of Huntington's Disease.J Huntingtons Dis. 2016 May 7;5(2):133-47. doi: 10.3233/JHD-150180. J Huntingtons Dis. 2016. PMID: 27163946
-
Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases.Int J Med Sci. 2019 Sep 20;16(10):1386-1396. doi: 10.7150/ijms.36516. eCollection 2019. Int J Med Sci. 2019. PMID: 31692944 Free PMC article. Review.
Cited by
-
The crucial role of vitamin C and its transporter (SVCT2) in bone marrow stromal cell autophagy and apoptosis.Stem Cell Res. 2015 Sep;15(2):312-21. doi: 10.1016/j.scr.2015.06.002. Epub 2015 Jun 10. Stem Cell Res. 2015. PMID: 26210298 Free PMC article.
-
Autophagy and neurodegeneration.J Clin Invest. 2015 Jan;125(1):65-74. doi: 10.1172/JCI73944. Epub 2015 Jan 2. J Clin Invest. 2015. PMID: 25654552 Free PMC article. Review.
-
Unbreak my heart: targeting mitochondrial autophagy in diabetic cardiomyopathy.Antioxid Redox Signal. 2015 Jun 10;22(17):1527-44. doi: 10.1089/ars.2015.6322. Epub 2015 Apr 28. Antioxid Redox Signal. 2015. PMID: 25808102 Free PMC article. Review.
-
Impaired Redox Signaling in Huntington's Disease: Therapeutic Implications.Front Mol Neurosci. 2019 Mar 19;12:68. doi: 10.3389/fnmol.2019.00068. eCollection 2019. Front Mol Neurosci. 2019. PMID: 30941013 Free PMC article. Review.
-
Medical relevance of UK-funded non-human primate research published from January 1997 to July 2012.J R Soc Med. 2014 Jul;107(7):264-270. doi: 10.1177/0141076814530686. Epub 2014 Apr 16. J R Soc Med. 2014. PMID: 24739383 Free PMC article. Review.
References
-
- Weintraub D., Comella C.L., Horn S. Parkinson's disease—Part 1: pathophysiology, symptoms, burden, diagnosis, and assessment. Am. J. Manag. Care. 2008;14:S40–S48. - PubMed
-
- Phillips W., Shannon K.M., Barker R.A. The current clinical management of Huntington's disease. Mov. Disord. 2008;23:1491–1504. doi:10.1002/mds.21971. - DOI - PubMed
-
- Stack E.C., Matson W.R., Ferrante R.J. Evidence of oxidant damage in Huntington's disease: translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 2008;1147:79–92. - PubMed
-
- Berger Z., Ravikumar B., Menzies F.M., Oroz L.G., Underwood B.R., Pangalos M.N., Schmitt I., Wullner U., Evert B.O., O'Kane C.J., et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006;15:433–442. doi:10.1093/hmg/ddi458. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
