

Innate retroviral restriction by Apobec3 promotes antibody affinity maturation in vivo
- PMID: 20566830
- PMCID: PMC3024598
- DOI: 10.4049/jimmunol.1001143
Innate retroviral restriction by Apobec3 promotes antibody affinity maturation in vivo
Abstract
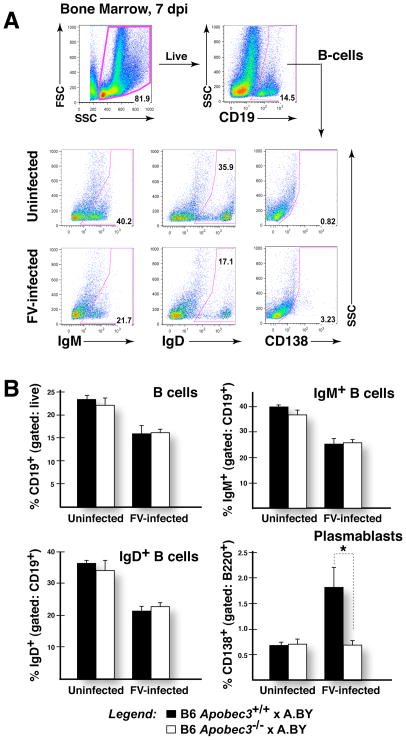
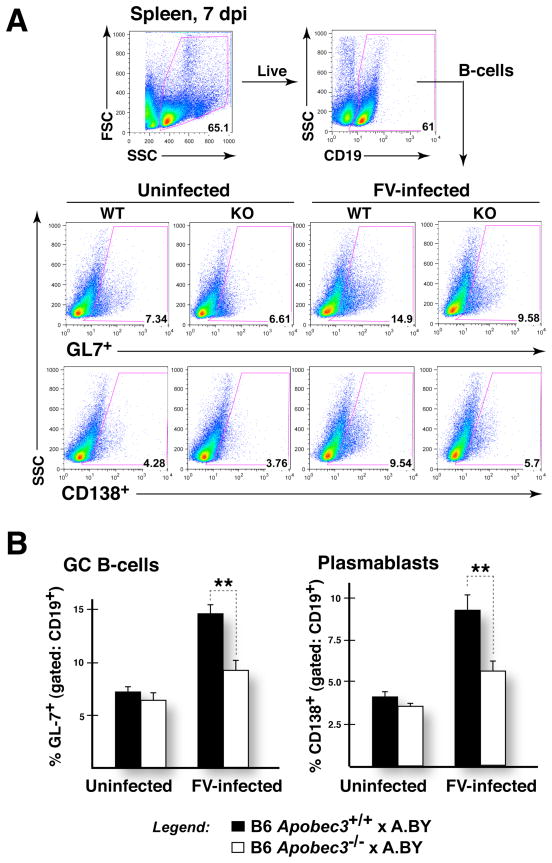
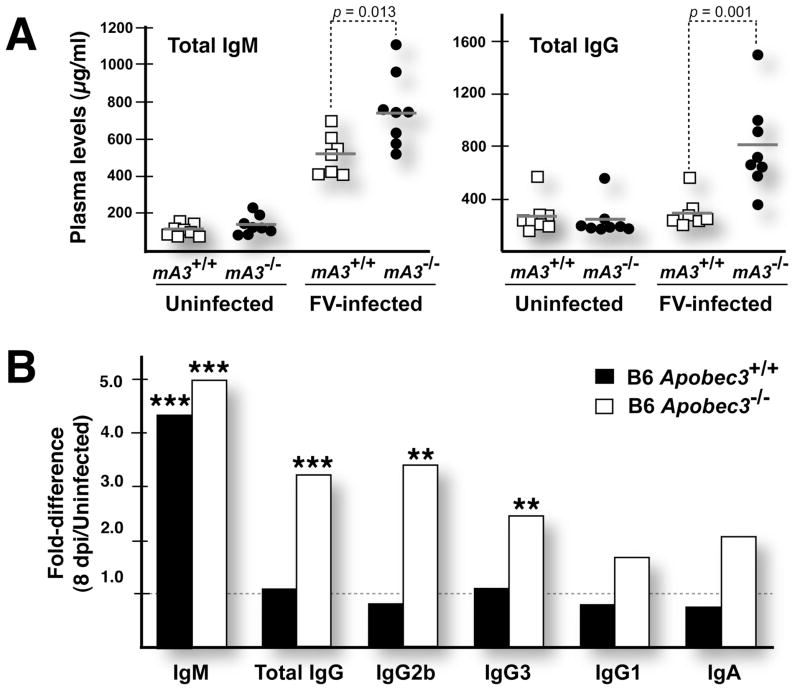
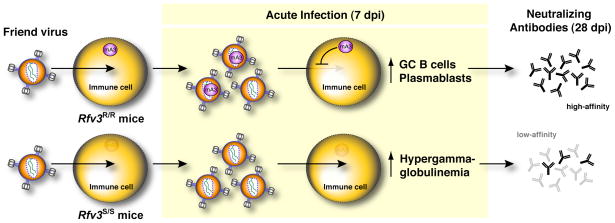
Apobec3/Rfv3 is an innate immune factor that promotes the neutralizing Ab response against Friend retrovirus (FV) in infected mice. Based on its evolutionary relationship to activation-induced deaminase, Apobec3 might directly influence Ab class switching and affinity maturation independently of viral infection. Alternatively, the antiviral activity of Apobec3 may indirectly influence neutralizing Ab responses by reducing early FV-induced pathology in critical immune compartments. To distinguish between these possibilities, we immunized wild-type and Apobec3-deficient C57BL/6 (B6) mice with (4-hydroxy-3-nitrophenyl) acetyl (NP) hapten and evaluated the binding affinity of the resultant NP-specific Abs. These studies revealed similar affinity maturation of NP-specific IgG1 Abs between wild-type and Apobec3-deficient mice in the absence of FV infection. In contrast, hapten-specific Ab affinity maturation was significantly compromised in Apobec3-deficient mice infected with FV. In highly susceptible (B6 x A.BY)F(1) mice, the B6 Apobec3 gene protected multiple cell types in the bone marrow and spleen from acute FV infection, including erythroid, B, T, and myeloid cells. In addition, B6 Apobec3 deficiency was associated with elevated Ig levels, but decreased induction of splenic germinal center B cells and plasmablasts during acute FV infection. These data suggest that Apobec3 indirectly influences FV-specific neutralizing Ab responses by reducing virus-induced immune dysfunction. These findings raise the possibility that enabling Apobec3 activity during acute infection with human pathogenic retroviruses, such as HIV-1, may similarly facilitate stronger virus-specific neutralizing Ab responses.
Figures
Similar articles
-
Persistence of viremia and production of neutralizing antibodies differentially regulated by polymorphic APOBEC3 and BAFF-R loci in friend virus-infected mice.J Virol. 2010 Jun;84(12):6082-95. doi: 10.1128/JVI.02516-09. Epub 2010 Apr 7. J Virol. 2010. PMID: 20375169 Free PMC article.
-
Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection.Science. 2008 Sep 5;321(5894):1343-6. doi: 10.1126/science.1161121. Science. 2008. PMID: 18772436 Free PMC article.
-
Immunoglobulin somatic hypermutation by APOBEC3/Rfv3 during retroviral infection.Proc Natl Acad Sci U S A. 2014 May 27;111(21):7759-64. doi: 10.1073/pnas.1403361111. Epub 2014 May 12. Proc Natl Acad Sci U S A. 2014. PMID: 24821801 Free PMC article.
-
Host genetic factors that control immune responses to retrovirus infections.Vaccine. 2008 Jun 6;26(24):2981-96. doi: 10.1016/j.vaccine.2008.01.004. Epub 2008 Jan 22. Vaccine. 2008. PMID: 18255203 Review.
-
AID for innate immunity to retroviral transformation.Immunity. 2006 Jun;24(6):671-672. doi: 10.1016/j.immuni.2006.06.005. Immunity. 2006. PMID: 16782023 Review.
Cited by
-
SAMHD1 Promotes the Antiretroviral Adaptive Immune Response in Mice Exposed to Lipopolysaccharide.J Immunol. 2022 Jan 15;208(2):444-453. doi: 10.4049/jimmunol.2001389. Epub 2021 Dec 10. J Immunol. 2022. PMID: 34893529 Free PMC article.
-
Type I interferon signaling is required for the APOBEC3/Rfv3-dependent neutralizing antibody response but not innate retrovirus restriction.Retrovirology. 2017 Apr 17;14(1):25. doi: 10.1186/s12977-017-0349-2. Retrovirology. 2017. PMID: 28415995 Free PMC article.
-
IFN-α treatment inhibits acute Friend retrovirus replication primarily through the antiviral effector molecule Apobec3.J Immunol. 2013 Feb 15;190(4):1583-90. doi: 10.4049/jimmunol.1202920. Epub 2013 Jan 11. J Immunol. 2013. PMID: 23315078 Free PMC article.
-
Ribonuclease L is not critical for innate restriction and adaptive immunity against Friend retrovirus infection.Virology. 2013 Aug 15;443(1):134-42. doi: 10.1016/j.virol.2013.05.009. Epub 2013 May 29. Virology. 2013. PMID: 23725696 Free PMC article.
-
Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1.Front Immunol. 2018 Dec 6;9:2876. doi: 10.3389/fimmu.2018.02876. eCollection 2018. Front Immunol. 2018. PMID: 30574147 Free PMC article. Review.
References
-
- Hasenkrug KJ, Dittmer U. Immune control and prevention of chronic Friend retrovirus infection. Front Biosci. 2007;12:1544–1551. - PubMed
-
- Miyazawa M, Tsuji-Kawahara S, Kanari Y. Host genetic factors that control immune responses to retrovirus infections. Vaccine. 2008;26:2981–2996. - PubMed
-
- Lilly F. Fv-2: identification and location of a second gene governing the spleen focus response to Friend leukemia virus in mice. Journal of the National Cancer Institute. 1970;45:163–169. - PubMed
-
- Persons DA, Paulson RF, Loyd MR, Herley MT, Bodner SM, Bernstein A, Correll PH, Ney PA. Fv2 encodes a truncated form of the Stk receptor tyrosine kinase. Nat Genet. 1999;23:159–165. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
