

(Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators
- PMID: 20568780
- PMCID: PMC2920492
- DOI: 10.1021/jm100217a
(Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators
Abstract
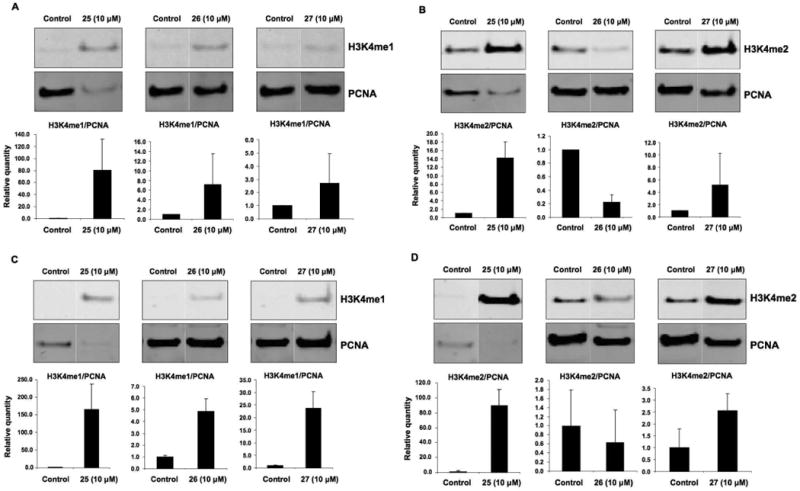
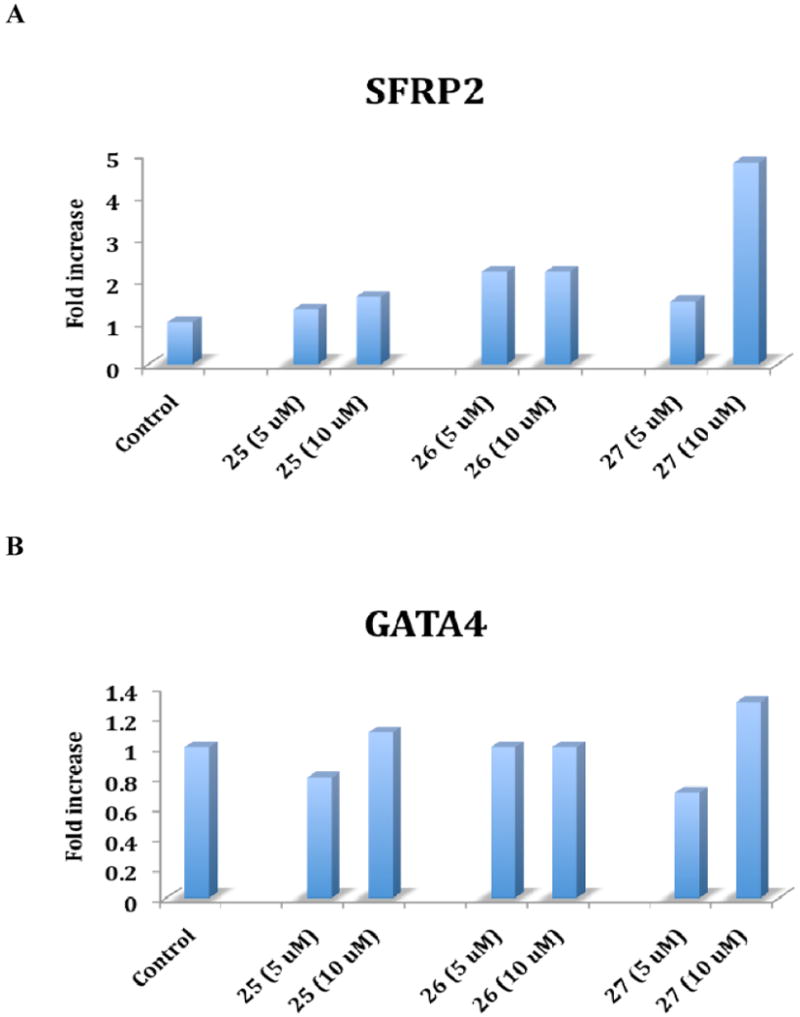
The recently discovered enzyme lysine-specific demethylase 1 (LSD1) plays an important role in the epigenetic control of gene expression, and aberrant gene silencing secondary to LSD1 overexpression is thought to contribute to the development of cancer. We recently reported a series of (bis)guanidines and (bis)biguanides that are potent inhibitors of LSD1 and induce the re-expression of aberrantly silenced tumor suppressor genes in tumor cells in vitro. We now report a series of isosteric ureas and thioureas that are also potent inhibitors of LSD1. These compounds induce increases in methylation at the histone 3 lysine 4 (H3K4) chromatin mark, a specific target of LSD1, in Calu-6 lung carcinoma cells. In addition, these analogues increase cellular levels of secreted frizzle-related protein (SFRP) 2 and transcription factor GATA4. These compounds represent an important new series of epigenetic modulators with the potential for use as antitumor agents.
Figures
Similar articles
-
Low molecular weight amidoximes that act as potent inhibitors of lysine-specific demethylase 1.J Med Chem. 2012 Sep 13;55(17):7378-91. doi: 10.1021/jm3002845. Epub 2012 Sep 4. J Med Chem. 2012. PMID: 22876979 Free PMC article.
-
Structure-activity study for (bis)ureidopropyl- and (bis)thioureidopropyldiamine LSD1 inhibitors with 3-5-3 and 3-6-3 carbon backbone architectures.Bioorg Med Chem. 2015 Apr 1;23(7):1601-12. doi: 10.1016/j.bmc.2015.01.049. Epub 2015 Feb 7. Bioorg Med Chem. 2015. PMID: 25725609 Free PMC article.
-
The re-expression of the epigenetically silenced e-cadherin gene by a polyamine analogue lysine-specific demethylase-1 (LSD1) inhibitor in human acute myeloid leukemia cell lines.Amino Acids. 2014 Mar;46(3):585-94. doi: 10.1007/s00726-013-1485-1. Epub 2013 Mar 19. Amino Acids. 2014. PMID: 23508577 Free PMC article.
-
Reversible Lysine Specific Demethylase 1 (LSD1) Inhibitors: A Promising Wrench to Impair LSD1.J Med Chem. 2021 Mar 11;64(5):2466-2488. doi: 10.1021/acs.jmedchem.0c02176. Epub 2021 Feb 23. J Med Chem. 2021. PMID: 33619958 Review.
-
Inhibitors of LSD1 as a potential therapy for acute myeloid leukemia.Expert Opin Investig Drugs. 2016 Jul;25(7):771-80. doi: 10.1080/13543784.2016.1175432. Epub 2016 Apr 21. Expert Opin Investig Drugs. 2016. PMID: 27077938 Review.
Cited by
-
Targeting the LSD1/KDM1 Family of Lysine Demethylases in Cancer and Other Human Diseases.Adv Exp Med Biol. 2023;1433:15-49. doi: 10.1007/978-3-031-38176-8_2. Adv Exp Med Biol. 2023. PMID: 37751134 Review.
-
Mechanochemical synthesis of thioureas, ureas and guanidines.Beilstein J Org Chem. 2017 Sep 1;13:1828-1849. doi: 10.3762/bjoc.13.178. eCollection 2017. Beilstein J Org Chem. 2017. PMID: 28904627 Free PMC article. Review.
-
Low molecular weight amidoximes that act as potent inhibitors of lysine-specific demethylase 1.J Med Chem. 2012 Sep 13;55(17):7378-91. doi: 10.1021/jm3002845. Epub 2012 Sep 4. J Med Chem. 2012. PMID: 22876979 Free PMC article.
-
KDM1 class flavin-dependent protein lysine demethylases.Biopolymers. 2015 Jul;104(4):213-46. doi: 10.1002/bip.22643. Biopolymers. 2015. PMID: 25787087 Free PMC article. Review.
-
Cyclic peptide inhibitors of lysine-specific demethylase 1 with improved potency identified by alanine scanning mutagenesis.Eur J Med Chem. 2018 Mar 25;148:210-220. doi: 10.1016/j.ejmech.2018.01.098. Epub 2018 Feb 7. Eur J Med Chem. 2018. PMID: 29459279 Free PMC article.
References
-
- Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13(6):477–483. - PubMed
-
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. - PubMed
-
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. - PubMed
-
- Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1(4):287–299. - PubMed
-
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Chemical Information
Miscellaneous
