

Long-term 12 year follow-up of X-linked congenital retinoschisis
- PMID: 20569020
- PMCID: PMC2997439
- DOI: 10.3109/13816810.2010.482555
Long-term 12 year follow-up of X-linked congenital retinoschisis
Abstract
Purpose: To investigate the retinal structure and function during the progression of X-linked retinoschisis (XLRS) from childhood to adulthood.
Methods: Ten patients clinically diagnosed with XLRS were investigated at 6-15 years of age (mean age 9 years) with a follow-up 8 to 14 years later (mean 12 years). The patients underwent regular ophthalmic examination as well as testing of best corrected visual acuity (BCVA), visual field (VF) and assessment of full-field electroretinography (ERG) during their first visit. During the follow-up, the same clinical protocols were repeated. In addition, macular structure and function was examined with multifocal electroretinography (mfERG) and optical coherence tomography (OCT). The patients were 18-25 years of age (mean age 21 years) at the follow-up examination. All exons and exon-intron boundaries of RS1-gene were sequenced for gene mutations in 9 out of the 10 patients.
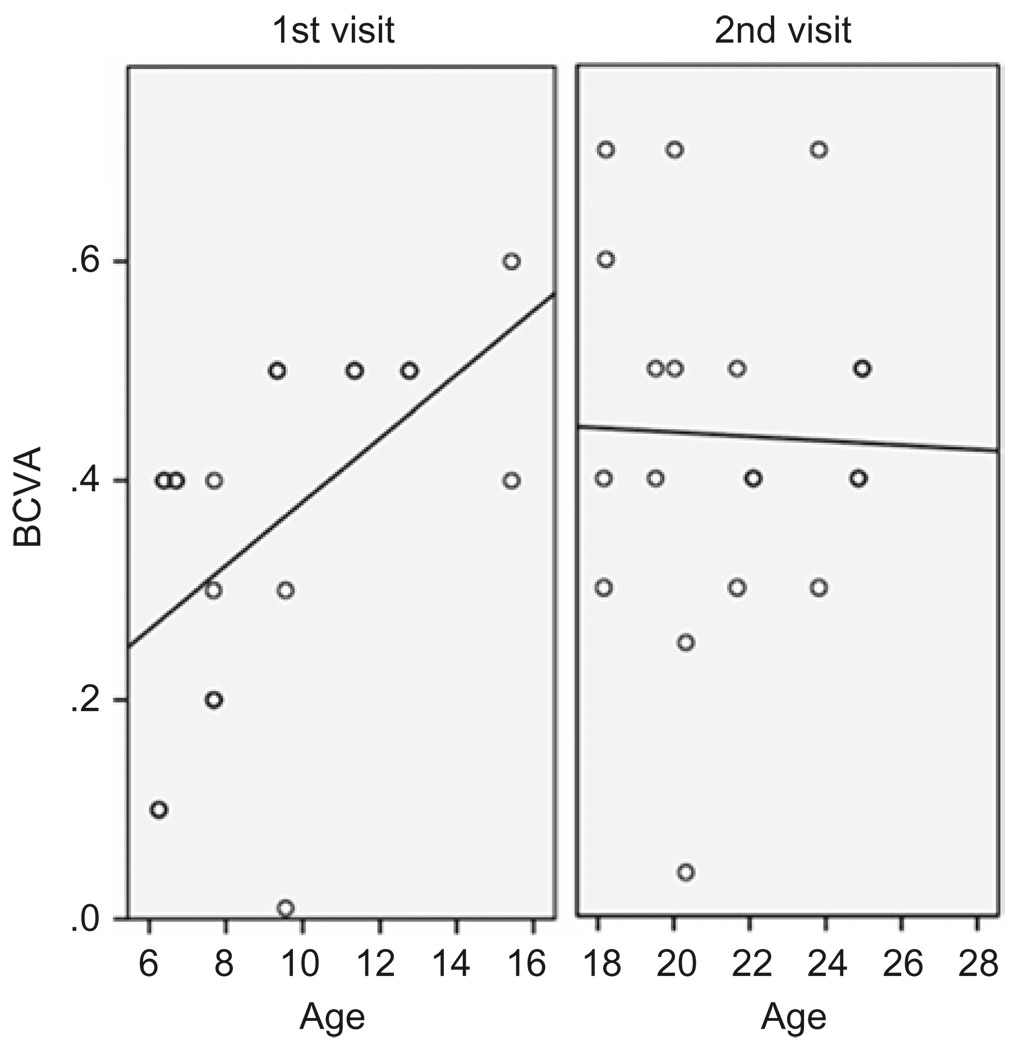
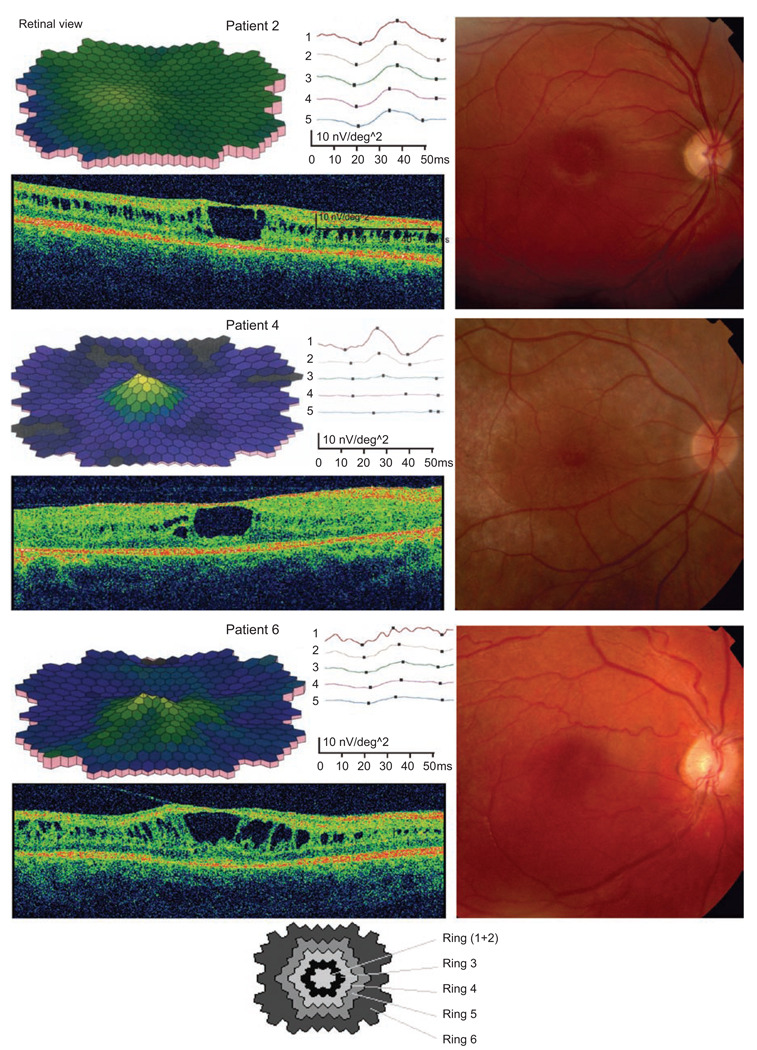
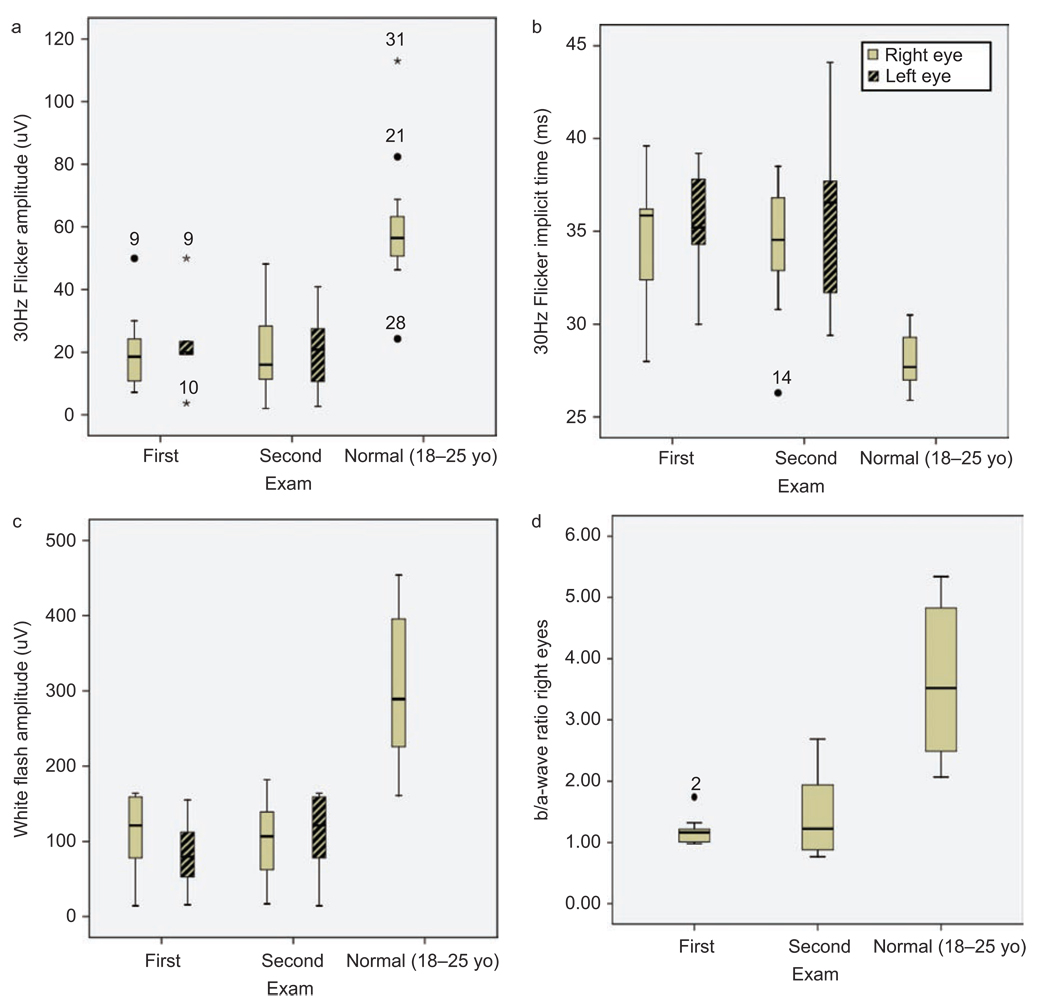
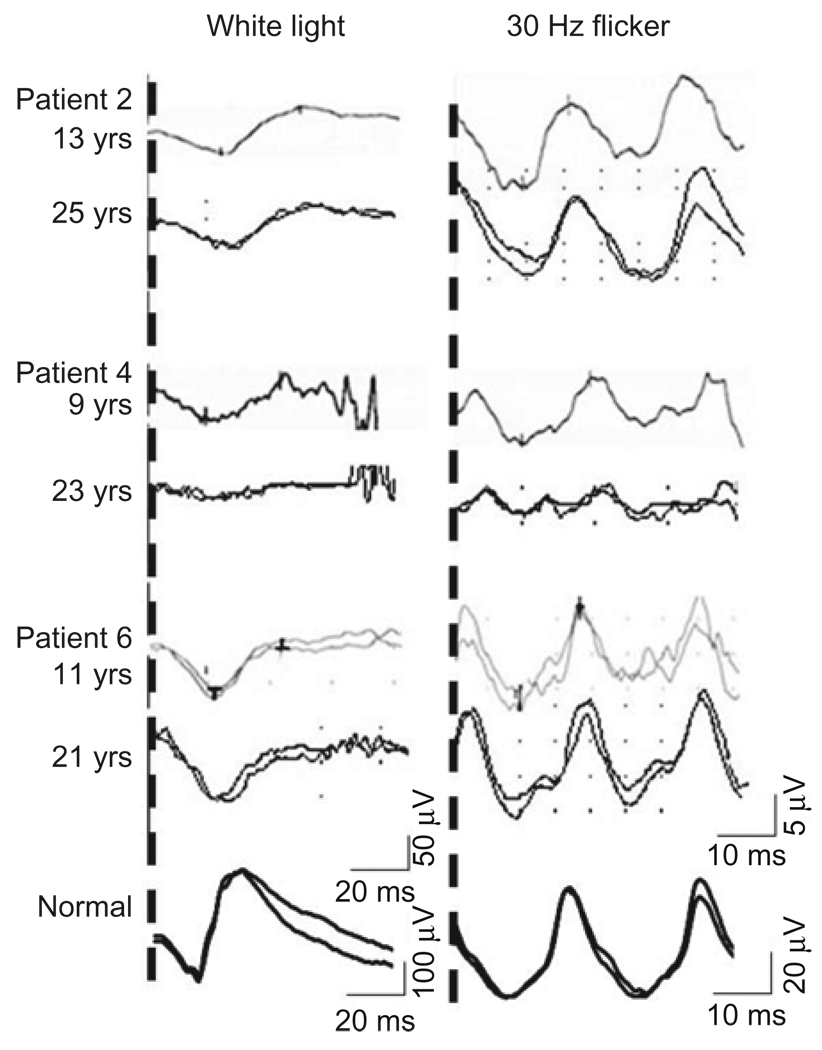
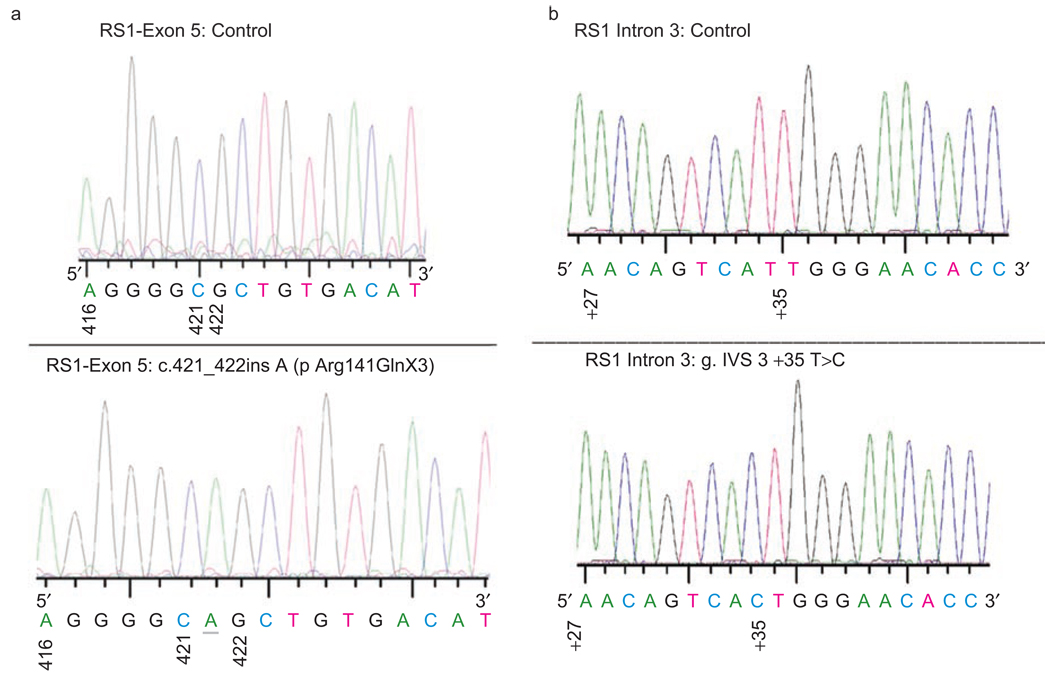
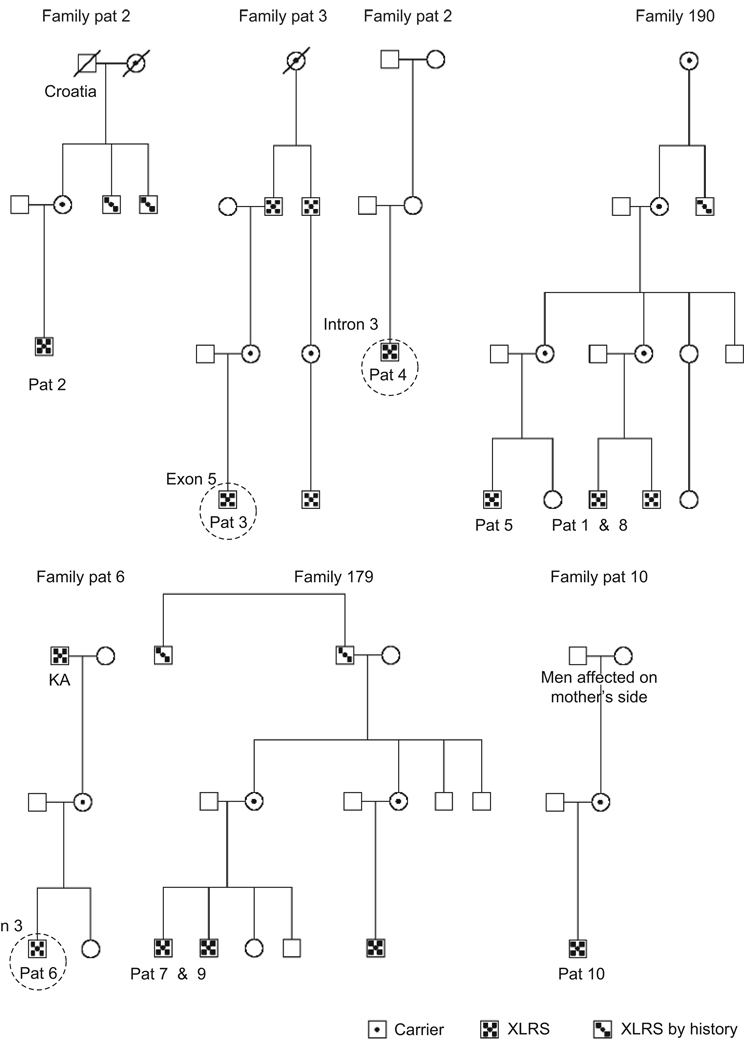
Results: Best corrected VA and VF were stable during this follow-up period. No significant progression in cone or rod function could be measured by full-field ERG. Multifocal electroretinography and OCT demonstrated a wide heterogeneity of macular changes in retinal structure and function at the time of follow-up visit. Three different mutations were detected in these nine patients, including a known nonsense mutation in exon 3, a novel insertion in exon 5 and an intronic mutation at 5' splice site of intron 3.
Conclusions: Clinical follow-up (mean 12 years) of ten young XLRS patients (mean age of 9 years) with a typical congenital retinoschisis phenotype revealed no significant decline in retinal function during this time period. MfERG and OCT demonstrated a wide variety of macular changes including structure and dysfunction. The XLRS disease was relatively stable during this period of observation and would afford opportunity for therapy studies to judge benefit against baseline and against the fellow eye.
Conflict of interest statement
Figures
Similar articles
-
Clinical and genetic findings in Hungarian patients with X-linked juvenile retinoschisis.Mol Vis. 2008;14:2321-32. Epub 2008 Dec 12. Mol Vis. 2008. PMID: 19093009 Free PMC article.
-
Abnormal cone structure in foveal schisis cavities in X-linked retinoschisis from mutations in exon 6 of the RS1 gene.Invest Ophthalmol Vis Sci. 2011 Dec 20;52(13):9614-23. doi: 10.1167/iovs.11-8600. Invest Ophthalmol Vis Sci. 2011. PMID: 22110067 Free PMC article.
-
Null retinoschisin-protein expression from an RS1 c354del1-ins18 mutation causing progressive and severe XLRS in a cross-sectional family study.Invest Ophthalmol Vis Sci. 2009 Nov;50(11):5375-83. doi: 10.1167/iovs.09-3839. Epub 2009 May 27. Invest Ophthalmol Vis Sci. 2009. PMID: 19474399 Free PMC article.
-
Of men and mice: Human X-linked retinoschisis and fidelity in mouse modeling.Prog Retin Eye Res. 2022 Mar;87:100999. doi: 10.1016/j.preteyeres.2021.100999. Epub 2021 Aug 11. Prog Retin Eye Res. 2022. PMID: 34390869 Review.
-
Review: Clinical findings and genetic characterization of children affected with X-linked retinoschisis in the Spanish population.Eur J Ophthalmol. 2025 May;35(3):809-820. doi: 10.1177/11206721241305244. Epub 2024 Dec 8. Eur J Ophthalmol. 2025. PMID: 39648411 Review.
Cited by
-
Luminance Thresholds and Their Correlation With Retinal Structure in X-Linked Retinoschisis.Invest Ophthalmol Vis Sci. 2021 Oct 4;62(13):25. doi: 10.1167/iovs.62.13.25. Invest Ophthalmol Vis Sci. 2021. PMID: 34705026 Free PMC article.
-
Structure/Psychophysical Relationships in X-Linked Retinoschisis.Invest Ophthalmol Vis Sci. 2016 Feb;57(2):332-7. doi: 10.1167/iovs.15-18354. Invest Ophthalmol Vis Sci. 2016. PMID: 26830370 Free PMC article.
-
Analysis of Anatomic and Functional Measures in X-Linked Retinoschisis.Invest Ophthalmol Vis Sci. 2018 Jun 1;59(7):2841-2847. doi: 10.1167/iovs.17-23297. Invest Ophthalmol Vis Sci. 2018. PMID: 30025115 Free PMC article.
-
Spatial and Temporal Integration Abnormalities in X-Linked Retinoschisis.Invest Ophthalmol Vis Sci. 2022 Aug 2;63(9):22. doi: 10.1167/iovs.63.9.22. Invest Ophthalmol Vis Sci. 2022. PMID: 35984651 Free PMC article.
-
"There Are Hills and Valleys": Experiences of Parenting a Son With X-Linked Retinoschisis.Am J Ophthalmol. 2020 Apr;212:98-104. doi: 10.1016/j.ajo.2019.11.023. Epub 2019 Nov 23. Am J Ophthalmol. 2020. PMID: 31765628 Free PMC article.
References
-
- Sauer CG, Gehrig A, Warneke-Wittstock R, et al. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nature Genetics. 1997;17:164–170. - PubMed
-
- The Retinoschisis C. Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. The Retinoschisis Consortium. Human Molecular Genetics. 1998;7:1185–1192. - PubMed
-
- Takada Y, Fariss RN, Tanikawa A, et al. A retinal neuronal developmental wave of retinoschisin expression begins in ganglion cells during layer formation. Investigative Ophthalmology & Visual Science. 2004;45:3302–3312. - PubMed
-
- Takada Y, Fariss RN, Muller M, et al. Retinoschisin expression and localization in rodent and human pineal and consequences of mouse RS1 gene knockout. Molecular Vision. 2006;12:1108–1116. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources