

Neonatal gene therapy of glycogen storage disease type Ia using a feline immunodeficiency virus-based vector
- PMID: 20571544
- PMCID: PMC2956916
- DOI: 10.1038/mt.2010.119
Neonatal gene therapy of glycogen storage disease type Ia using a feline immunodeficiency virus-based vector
Abstract
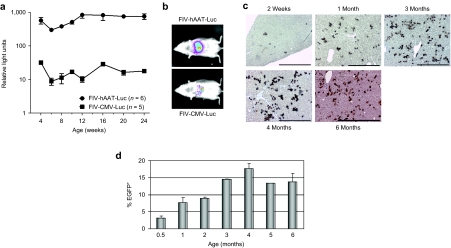
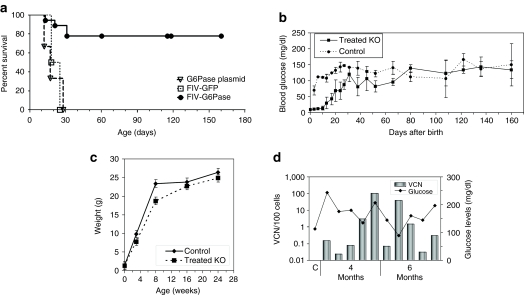
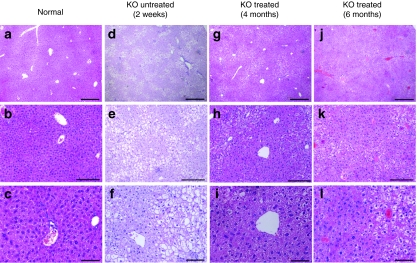
Glycogen storage disease type Ia (GSD-Ia), also known as von Gierke disease, is caused by a deficiency of glucose-6-phosphatase-alpha (G6Pase), a key enzyme in glucose homeostasis. From birth, affected individuals cannot maintain normal blood glucose levels and suffer from a variety of metabolic disorders, leading to life-threatening complications. Gene therapy has been proposed as a possible option for treatment of this illness. Vectors have been constructed from feline immunodeficiency virus (FIV), a nonprimate lentivirus, because the wild-type virus does not cause disease in humans. Previously, we have shown that these vectors are capable of integrating stably into hepatocyte cell lines and adult murine livers and lead to long-term transgene expression. In the current work, we have assessed the ability to attenuate disease symptoms in a murine model of GSD-Ia. Single administration of FIV vectors containing the human G6Pase gene to G6Pase-alpha(-/-) mice did not change the biochemical and pathological phenotype. However, a double neonatal administration protocol led to normalized blood glucose levels, significantly extended survival, improved body weight, and decreased accumulation of liver glycogen associated with the disease. This approach shows a promising paradigm for treating GSD-Ia patients early in life thereby avoiding long-term consequences.
Figures

Similar articles
-
Efficacy of helper-dependent adenovirus vector-mediated gene therapy in murine glycogen storage disease type Ia.Mol Ther. 2007 Jul;15(7):1253-8. doi: 10.1038/sj.mt.6300188. Epub 2007 May 15. Mol Ther. 2007. PMID: 17505475
-
Early, sustained efficacy of adeno-associated virus vector-mediated gene therapy in glycogen storage disease type Ia.Gene Ther. 2006 Sep;13(17):1281-9. doi: 10.1038/sj.gt.3302774. Epub 2006 May 4. Gene Ther. 2006. PMID: 16672983
-
Long-term correction of murine glycogen storage disease type Ia by recombinant adeno-associated virus-1-mediated gene transfer.Gene Ther. 2006 Feb;13(4):321-9. doi: 10.1038/sj.gt.3302650. Gene Ther. 2006. PMID: 16195703
-
Emerging roles of autophagy in hepatic tumorigenesis and therapeutic strategies in glycogen storage disease type Ia: A review.J Inherit Metab Dis. 2021 Jan;44(1):118-128. doi: 10.1002/jimd.12267. Epub 2020 Jul 2. J Inherit Metab Dis. 2021. PMID: 32474930 Review.
-
Gene therapy for type I glycogen storage diseases.Curr Gene Ther. 2007 Apr;7(2):79-88. doi: 10.2174/156652307780363152. Curr Gene Ther. 2007. PMID: 17430128 Free PMC article. Review.
Cited by
-
Altering α-dystroglycan receptor affinity of LCMV pseudotyped lentivirus yields unique cell and tissue tropism.Genet Vaccines Ther. 2011 Apr 8;9:8. doi: 10.1186/1479-0556-9-8. Genet Vaccines Ther. 2011. PMID: 21477292 Free PMC article.
-
Pseudotyped Viruses: A Useful Platform for Pre-Clinical Studies Conducted in a BSL-2 Laboratory Setting.Biomolecules. 2025 Jan 15;15(1):135. doi: 10.3390/biom15010135. Biomolecules. 2025. PMID: 39858529 Free PMC article. Review.
-
Hepatorenal correction in murine glycogen storage disease type I with a double-stranded adeno-associated virus vector.Mol Ther. 2011 Nov;19(11):1961-70. doi: 10.1038/mt.2011.126. Epub 2011 Jul 5. Mol Ther. 2011. PMID: 21730973 Free PMC article.
-
Gene therapy for glycogen storage diseases.J Inherit Metab Dis. 2024 Jan;47(1):93-118. doi: 10.1002/jimd.12654. Epub 2023 Jul 27. J Inherit Metab Dis. 2024. PMID: 37421310 Free PMC article. Review.
-
Gene transfer to chicks using lentiviral vectors administered via the embryonic chorioallantoic membrane.PLoS One. 2012;7(5):e36531. doi: 10.1371/journal.pone.0036531. Epub 2012 May 11. PLoS One. 2012. PMID: 22606269 Free PMC article.
References
-
- Chou JY. The molecular basis of type 1 glycogen storage diseases. Curr Mol Med. 2001;1:25–44. - PubMed
-
- Lei KJ, Chen H, Pan CJ, Ward JM, Mosinger B, Jr, Lee EJ, et al. Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type-1a mouse. Nat Genet. 1996;13:203–209. - PubMed
-
- Kishnani PS, Faulkner E, VanCamp S, Jackson M, Brown T, Boney A, et al. Canine model and genomic structural organization of glycogen storage disease type Ia (GSD Ia) Vet Pathol. 2001;38:83–91. - PubMed
-
- Ghosh A, Allamarvdasht M, Pan CJ, Sun MS, Mansfield BC, Byrne BJ, et al. Long-term correction of murine glycogen storage disease type Ia by recombinant adeno-associated virus-1-mediated gene transfer. Gene Ther. 2006;13:321–329. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
