

Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy
- PMID: 20573906
- PMCID: PMC6634618
- DOI: 10.1523/JNEUROSCI.0633-10.2010
Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy
Abstract
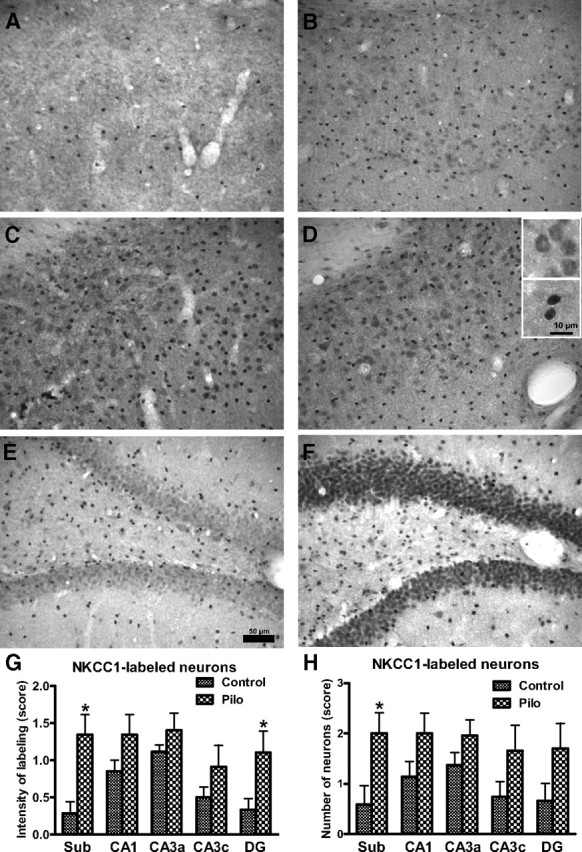
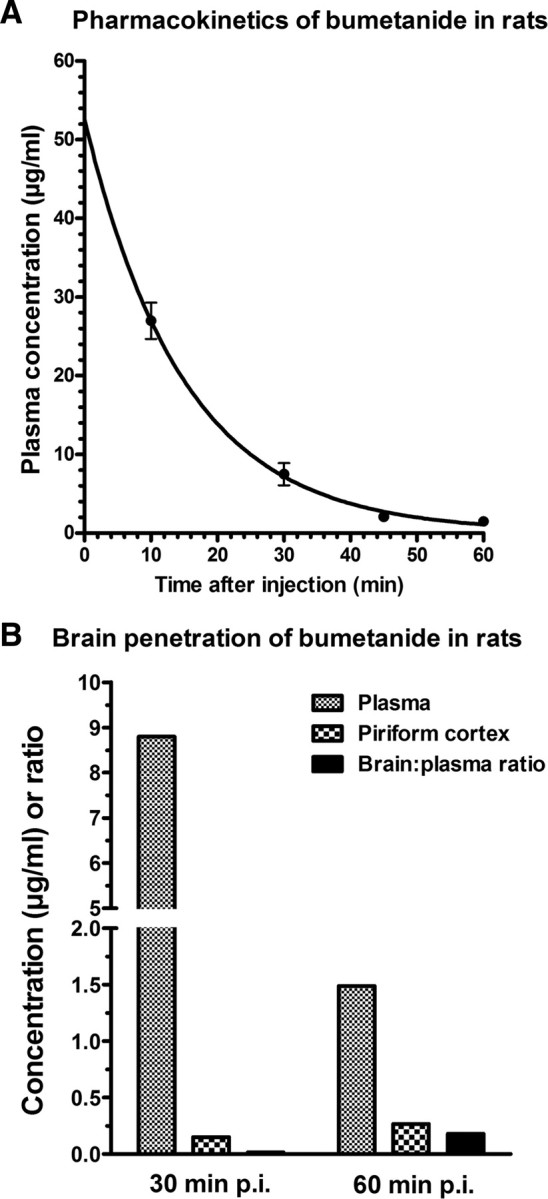
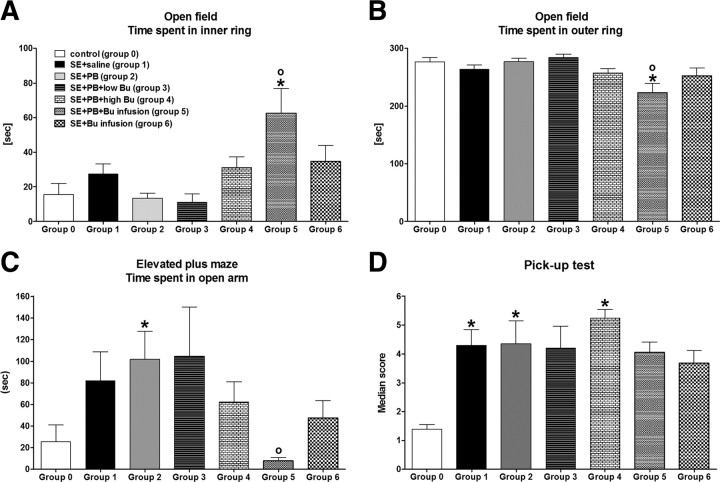
Accumulating evidence suggests that changes in neuronal chloride homeostasis may be involved in the mechanisms by which brain insults induce the development of epilepsy. A variety of brain insults, including status epilepticus (SE), lead to changes in the expression of the cation-chloride cotransporters KCC2 and NKCC1, resulting in intracellular chloride accumulation and reappearance of immature, depolarizing synaptic responses to GABA(A) receptor activation, which may critically contribute to the neuronal hyperexcitability underlying epileptogenesis. In the present study, it was evaluated whether prolonged administration of the selective NKCC1 inhibitor, bumetanide, after a pilocarpine-induced SE modifies the development of epilepsy in adult female rats. The antiepileptic drug phenobarbital, either alone or in combination, was used for comparison. Based on pharmacokinetic studies with bumetanide, which showed extremely rapid elimination and low brain penetration of this drug in rats, bumetanide was administered systemically with different dosing protocols, including continuous intravenous infusion. As shown by immunohistochemistry, neuronal NKCC1 expression was markedly upregulated shortly after SE. Prophylactic treatment with phenobarbital after SE reduced the number of rats developing spontaneous seizures and decreased seizure frequency, indicating a disease-modifying effect. Bumetanide did not exert any significant effects on development of spontaneous seizures nor did it enhance the effects of phenobarbital. However, combined treatment with both drugs counteracted several of the behavioral consequences of SE, which was not observed with single drug treatment. These data do not indicate that bumetanide can prevent epilepsy after SE, but the disease-modifying effect of this drug warrants further studies with more lipophilic prodrugs of bumetanide.
Figures


References
-
- Bankstahl JP, Löscher W. Resistance to antiepileptic drugs and expression of P-glycoprotein in two rat models of status epilepticus. Epilepsy Res. 2008;82:70–85. - PubMed
-
- Bankstahl JP, Hoffmann K, Bethmann K, Löscher W. Glutamate is critically involved in seizure-induced overexpression of P-glycoprotein in the brain. Neuropharmacology. 2008;54:1006–1016. - PubMed
-
- Baulac M. Phenobarbital and other barbiturates: clinical efficacy and use in epilepsy. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic drugs. Ed 5. Philadelphia: Lippincott Williams and Wilkins; 2002. pp. 514–521.
-
- Ben-Ari Y, Holmes GL. The multiple facets of gamma-aminobutyric acid dysfunction in epilepsy. Curr Opin Neurol. 2005;18:141–145. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials