

X-linked cone dystrophy caused by mutation of the red and green cone opsins
- PMID: 20579627
- PMCID: PMC2896775
- DOI: 10.1016/j.ajhg.2010.05.019
X-linked cone dystrophy caused by mutation of the red and green cone opsins
Abstract
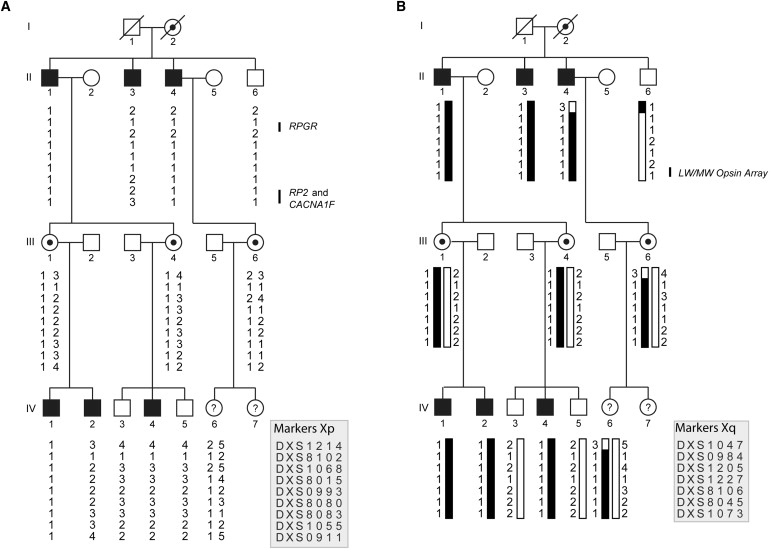
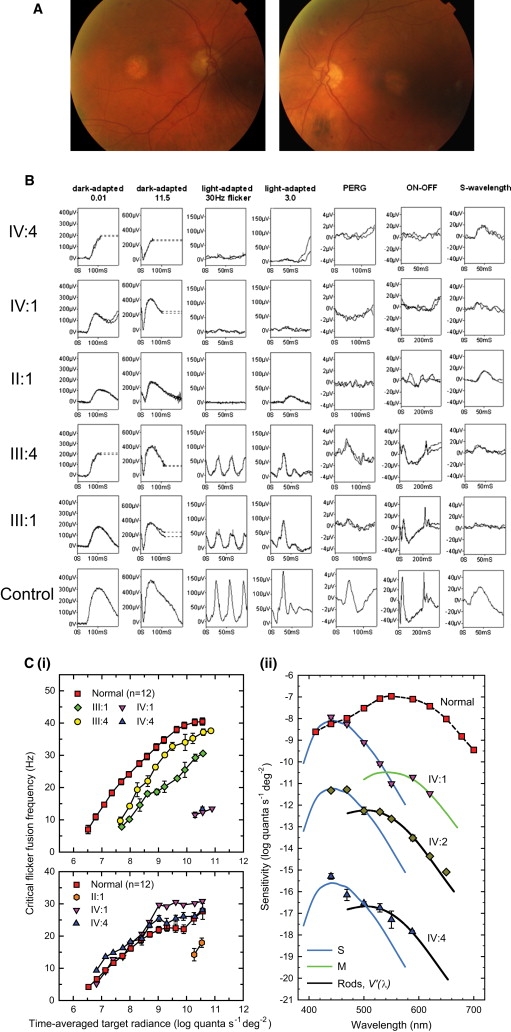
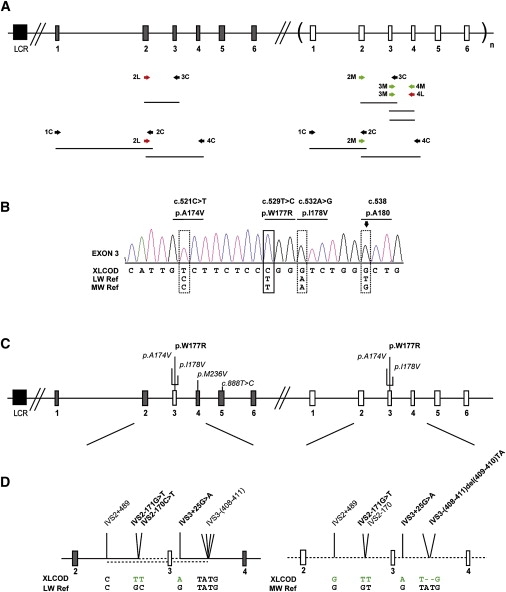
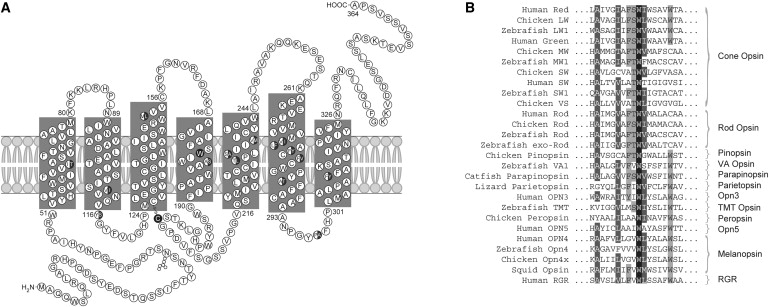
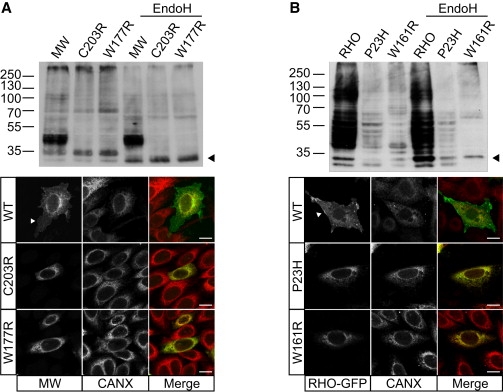
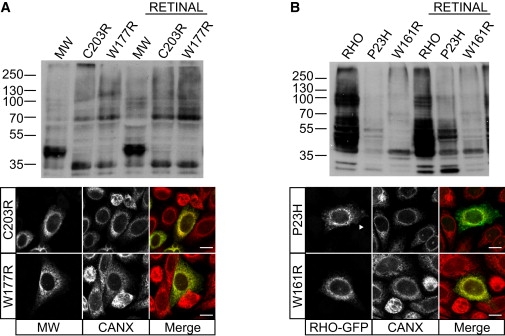
X-linked cone and cone-rod dystrophies (XLCOD and XLCORD) are a heterogeneous group of progressive disorders that solely or primarily affect cone photoreceptors. Mutations in exon ORF15 of the RPGR gene are the most common underlying cause. In a previous study, we excluded RPGR exon ORF15 in some families with XLCOD. Here, we report genetic mapping of XLCOD to Xq26.1-qter. A significant LOD score was detected with marker DXS8045 (Z(max) = 2.41 [theta = 0.0]). The disease locus encompasses the cone opsin gene array on Xq28. Analysis of the array revealed a missense mutation (c. 529T>C [p. W177R]) in exon 3 of both the long-wavelength-sensitive (LW, red) and medium-wavelength-sensitive (MW, green) cone opsin genes that segregated with disease. Both exon 3 sequences were identical and were derived from the MW gene as a result of gene conversion. The amino acid W177 is highly conserved in visual and nonvisual opsins across species. We show that W177R in MW opsin and the equivalent W161R mutation in rod opsin result in protein misfolding and retention in the endoplasmic reticulum. We also demonstrate that W177R misfolding, unlike the P23H mutation in rod opsin that causes retinitis pigmentosa, is not rescued by treatment with the pharmacological chaperone 9-cis-retinal. Mutations in the LW/MW cone opsin gene array can, therefore, lead to a spectrum of disease, ranging from color blindness to progressive cone dystrophy (XLCOD5).
Copyright 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Michaelides M., Hardcastle A.J., Hunt D.M., Moore A.T. Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 2006;51:232–258. - PubMed
-
- Yang Z., Peachey N.S., Moshfeghi D.M., Thirumalaichary S., Chorich L., Shugart Y.Y., Fan K., Zhang K. Mutations in the RPGR gene cause X-linked cone dystrophy. Hum. Mol. Genet. 2002;11:605–611. - PubMed
-
- Ebenezer N.D., Michaelides M., Jenkins S.A., Audo I., Webster A.R., Cheetham M.E., Stockman A., Maher E.R., Ainsworth J.R., Yates J.R. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest. Ophthalmol. Vis. Sci. 2005;46:1891–1898. - PubMed
-
- Ruddle J.B., Ebenezer N.D., Kearns L.S., Mulhall L.E., Mackey D.A., Hardcastle A.J. RPGR ORF15 genotype and clinical variability of retinal degeneration in an Australian population. Br. J. Ophthalmol. 2009;93:1151–1154. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
