

A probabilistic and continuous model of protein conformational space for template-free modeling
- PMID: 20583926
- PMCID: PMC3203516
- DOI: 10.1089/cmb.2009.0235
A probabilistic and continuous model of protein conformational space for template-free modeling
Abstract
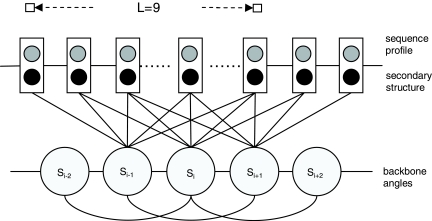
One of the major challenges with protein template-free modeling is an efficient sampling algorithm that can explore a huge conformation space quickly. The popular fragment assembly method constructs a conformation by stringing together short fragments extracted from the Protein Data Base (PDB). The discrete nature of this method may limit generated conformations to a subspace in which the native fold does not belong. Another worry is that a protein with really new fold may contain some fragments not in the PDB. This article presents a probabilistic model of protein conformational space to overcome the above two limitations. This probabilistic model employs directional statistics to model the distribution of backbone angles and 2(nd)-order Conditional Random Fields (CRFs) to describe sequence-angle relationship. Using this probabilistic model, we can sample protein conformations in a continuous space, as opposed to the widely used fragment assembly and lattice model methods that work in a discrete space. We show that when coupled with a simple energy function, this probabilistic method compares favorably with the fragment assembly method in the blind CASP8 evaluation, especially on alpha or small beta proteins. To our knowledge, this is the first probabilistic method that can search conformations in a continuous space and achieves favorable performance. Our method also generated three-dimensional (3D) models better than template-based methods for a couple of CASP8 hard targets. The method described in this article can also be applied to protein loop modeling, model refinement, and even RNA tertiary structure prediction.
Figures
Similar articles
-
Fragment-free approach to protein folding using conditional neural fields.Bioinformatics. 2010 Jun 15;26(12):i310-7. doi: 10.1093/bioinformatics/btq193. Bioinformatics. 2010. PMID: 20529922 Free PMC article.
-
A probabilistic model of RNA conformational space.PLoS Comput Biol. 2009 Jun;5(6):e1000406. doi: 10.1371/journal.pcbi.1000406. Epub 2009 Jun 19. PLoS Comput Biol. 2009. PMID: 19543381 Free PMC article.
-
A Probabilistic Graphical Model for Ab Initio Folding.Res Comput Mol Biol. 2009;5541:59-73. doi: 10.1007/978-3-642-02008-7_5. Res Comput Mol Biol. 2009. PMID: 23459639 Free PMC article.
-
A guide to template based structure prediction.Curr Protein Pept Sci. 2009 Jun;10(3):270-85. doi: 10.2174/138920309788452182. Curr Protein Pept Sci. 2009. PMID: 19519455 Review.
-
Progress and challenges in protein structure prediction.Curr Opin Struct Biol. 2008 Jun;18(3):342-8. doi: 10.1016/j.sbi.2008.02.004. Epub 2008 Apr 22. Curr Opin Struct Biol. 2008. PMID: 18436442 Free PMC article. Review.
Cited by
-
TASSER_WT: a protein structure prediction algorithm with accurate predicted contact restraints for difficult protein targets.Biophys J. 2010 Nov 3;99(9):3066-75. doi: 10.1016/j.bpj.2010.09.007. Biophys J. 2010. PMID: 21044605 Free PMC article.
-
Potentials of mean force for protein structure prediction vindicated, formalized and generalized.PLoS One. 2010 Nov 10;5(11):e13714. doi: 10.1371/journal.pone.0013714. PLoS One. 2010. PMID: 21103041 Free PMC article.
-
Protein Structure Classification and Loop Modeling Using Multiple Ramachandran Distributions.Comput Struct Biotechnol J. 2017 Feb 8;15:243-254. doi: 10.1016/j.csbj.2017.01.011. eCollection 2017. Comput Struct Biotechnol J. 2017. PMID: 28280526 Free PMC article.
-
Including residual contact information into replica-exchange MD simulations significantly enriches native-like conformations.PLoS One. 2020 Nov 16;15(11):e0242072. doi: 10.1371/journal.pone.0242072. eCollection 2020. PLoS One. 2020. PMID: 33196676 Free PMC article.
-
Assessing protein conformational sampling methods based on bivariate lag-distributions of backbone angles.Brief Bioinform. 2013 Nov;14(6):724-36. doi: 10.1093/bib/bbs052. Epub 2012 Aug 27. Brief Bioinform. 2013. PMID: 22926831 Free PMC article.
References
-
- Aarts E. Korst J. Simulated Annealing and Boltzmann Machines: A Stochastic Approach to Combinatorial Optimization and Neural Computing. Wiley; New York: 1991.
-
- Branden C.-I. Tooze J. Introduction to Protein Structure. Garland Publishing; New York: 1999.
-
- Claessens M. van Cutsem E. Lasters I., et al. Modelling the polypeptide backbone with “spare parts” from known protein structures. Protein Eng. 1989;2:335–345. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
