

Targeted genome-wide enrichment of functional regions
- PMID: 20585402
- PMCID: PMC2886846
- DOI: 10.1371/journal.pone.0011138
Targeted genome-wide enrichment of functional regions
Abstract
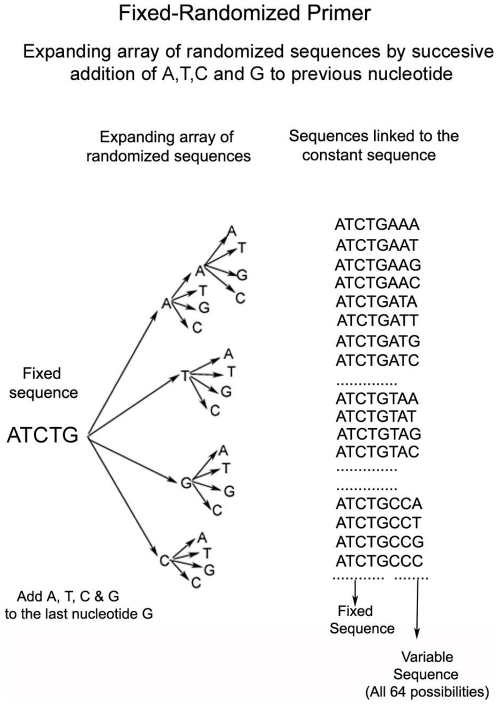
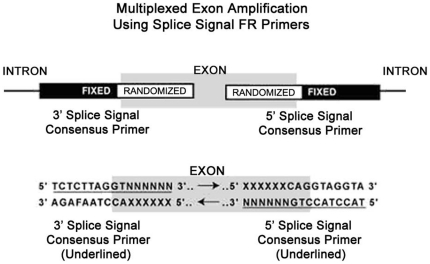
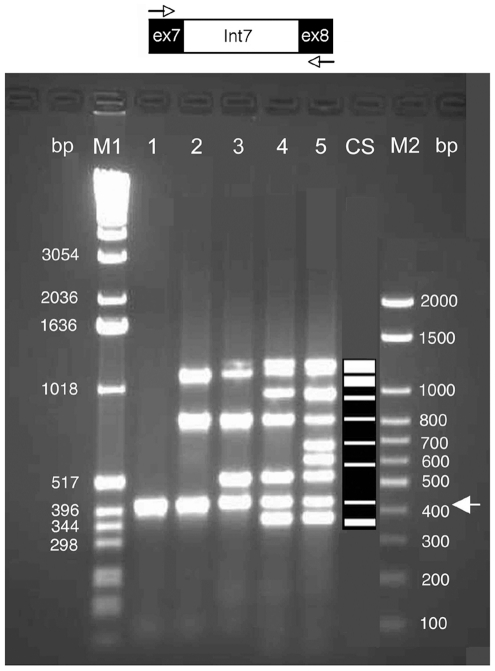
Only a small fraction of large genomes such as that of the human contains the functional regions such as the exons, promoters, and polyA sites. A platform technique for selective enrichment of functional genomic regions will enable several next-generation sequencing applications that include the discovery of causal mutations for disease and drug response. Here, we describe a powerful platform technique, termed "functional genomic fingerprinting" (FGF), for the multiplexed genomewide isolation and analysis of targeted regions such as the exome, promoterome, or exon splice enhancers. The technique employs a fixed part of a uniquely designed Fixed-Randomized primer, while the randomized part contains all the possible sequence permutations. The Fixed-Randomized primers bind with full sequence complementarity at multiple sites where the fixed sequence (such as the splice signals) occurs within the genome, and multiplex amplify many regions bounded by the fixed sequences (e.g., exons). Notably, validation of this technique using cardiac myosin binding protein-C (MYBPC3) gene as an example strongly supports the application and efficacy of this method. Further, assisted by genomewide computational analyses of such sequences, the FGF technique may provide a unique platform for high-throughput sample production and analysis of targeted genomic regions by the next-generation sequencing techniques, with powerful applications in discovering disease and drug response genes.
Conflict of interest statement
Figures

Similar articles
-
MPD: multiplex primer design for next-generation targeted sequencing.BMC Bioinformatics. 2017 Jan 5;18(1):14. doi: 10.1186/s12859-016-1453-3. BMC Bioinformatics. 2017. PMID: 28056760 Free PMC article.
-
Structural analysis of the human RFC-1 gene encoding a folate transporter reveals multiple promoters and alternatively spliced transcripts with 5' end heterogeneity.Gene. 1998 May 12;211(2):331-41. doi: 10.1016/s0378-1119(98)00123-1. Gene. 1998. PMID: 9602167
-
[Analysis, identification and correction of some errors of model refseqs appeared in NCBI Human Gene Database by in silico cloning and experimental verification of novel human genes].Yi Chuan Xue Bao. 2004 May;31(5):431-43. Yi Chuan Xue Bao. 2004. PMID: 15478601 Chinese.
-
Computational methods for the identification of genes in vertebrate genomic sequences.Hum Mol Genet. 1997;6(10):1735-44. doi: 10.1093/hmg/6.10.1735. Hum Mol Genet. 1997. PMID: 9300666 Review.
-
Mutations that alter RNA splicing of the human HPRT gene: a review of the spectrum.Mutat Res. 1998 Nov;411(3):179-214. doi: 10.1016/s1383-5742(98)00013-1. Mutat Res. 1998. PMID: 9804951 Review.
Cited by
-
Microbial Ecology: Where are we now?Postdoc J. 2016 Nov;4(11):3-17. doi: 10.14304/SURYA.JPR.V4N11.2. Postdoc J. 2016. PMID: 27975077 Free PMC article.
-
The impact of next-generation sequencing on genomics.J Genet Genomics. 2011 Mar 20;38(3):95-109. doi: 10.1016/j.jgg.2011.02.003. Epub 2011 Mar 15. J Genet Genomics. 2011. PMID: 21477781 Free PMC article. Review.
-
From genetics to genomics of epilepsy.Neurol Res Int. 2012;2012:876234. doi: 10.1155/2012/876234. Epub 2012 May 8. Neurol Res Int. 2012. PMID: 22645681 Free PMC article.
-
Applications of high-throughput DNA sequencing to benign hematology.Blood. 2013 Nov 21;122(22):3575-82. doi: 10.1182/blood-2013-07-460337. Epub 2013 Sep 10. Blood. 2013. PMID: 24021670 Free PMC article. Review.
-
Plant phylogenomics based on genome-partitioning strategies: Progress and prospects.Plant Divers. 2018 Jun 30;40(4):158-164. doi: 10.1016/j.pld.2018.06.005. eCollection 2018 Aug. Plant Divers. 2018. PMID: 30740560 Free PMC article. Review.
References
-
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. - PubMed
-
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. - PubMed
-
- Aparicio AJRS, Huntsman DG. Does massively parallel DNA resequencing signify the end of histopathology as we know it? J Pathol 2010. 2010;220:307–315. - PubMed
-
- Forrest AR, Carninci P. Whole genome transcriptome analysis. RNA Biol. 2009;6:107–12. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
