

Alterations in striatal synaptic transmission are consistent across genetic mouse models of Huntington's disease
- PMID: 20585470
- PMCID: PMC2888168
- DOI: 10.1042/AN20100007
Alterations in striatal synaptic transmission are consistent across genetic mouse models of Huntington's disease
Abstract
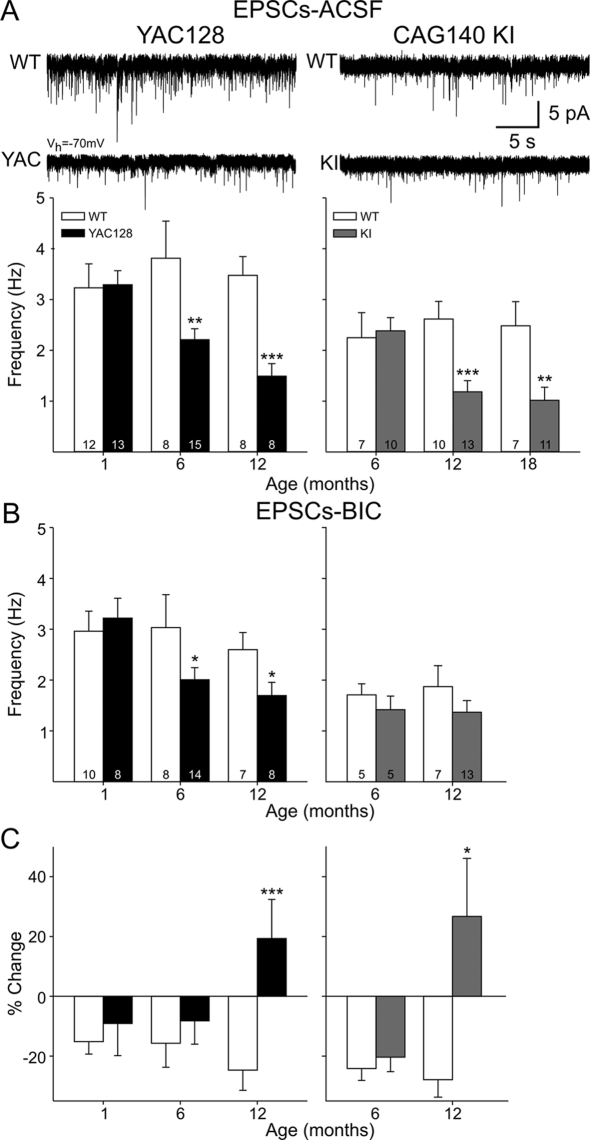
Since the identification of the gene responsible for HD (Huntington's disease), many genetic mouse models have been generated. Each employs a unique approach for delivery of the mutated gene and has a different CAG repeat length and background strain. The resultant diversity in the genetic context and phenotypes of these models has led to extensive debate regarding the relevance of each model to the human disorder. Here, we compare and contrast the striatal synaptic phenotypes of two models of HD, namely the YAC128 mouse, which carries the full-length huntingtin gene on a yeast artificial chromosome, and the CAG140 KI (knock-in) mouse, which carries a human/mouse chimaeric gene that is expressed in the context of the mouse genome, with our previously published data obtained from the R6/2 mouse, which is transgenic for exon 1 mutant huntingtin. We show that striatal MSNs (medium-sized spiny neurons) in YAC128 and CAG140 KI mice have similar electrophysiological phenotypes to that of the R6/2 mouse. These include a progressive increase in membrane input resistance, a reduction in membrane capacitance, a lower frequency of spontaneous excitatory postsynaptic currents and a greater frequency of spontaneous inhibitory postsynaptic currents in a subpopulation of striatal neurons. Thus, despite differences in the context of the inserted gene between these three models of HD, the primary electrophysiological changes observed in striatal MSNs are consistent. The outcomes suggest that the changes are due to the expression of mutant huntingtin and such alterations can be extended to the human condition.
Keywords: ACSF, artificial cerebrospinal fluid; AP5, dl-2-amino-5-phosphonovaleric acid; BIC, bicuculline methobromide; CAG 140 knock-in mouse model; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione; EPSC, excitatory postsynaptic current; GABAA, γ-aminobutyric acid type A; HD, Huntington's disease; HF, high frequency; Huntington's disease; IPSC, inhibitory postsynaptic current; KI, knock-in; LF, low frequency; MSN, medium-sized spiny neuron; WT, wild-type; YAC128 mouse model; electrophysiology; striatum.
Figures



Similar articles
-
Altered excitatory and inhibitory inputs to striatal medium-sized spiny neurons and cortical pyramidal neurons in the Q175 mouse model of Huntington's disease.J Neurophysiol. 2015 Apr 1;113(7):2953-66. doi: 10.1152/jn.01056.2014. Epub 2015 Feb 11. J Neurophysiol. 2015. PMID: 25673747 Free PMC article.
-
Striatal Direct and Indirect Pathway Output Structures Are Differentially Altered in Mouse Models of Huntington's Disease.J Neurosci. 2018 May 16;38(20):4678-4694. doi: 10.1523/JNEUROSCI.0434-18.2018. Epub 2018 Apr 24. J Neurosci. 2018. PMID: 29691329 Free PMC article.
-
Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington's disease.Neurobiol Dis. 2008 Jul;31(1):80-8. doi: 10.1016/j.nbd.2008.03.010. Epub 2008 Apr 16. Neurobiol Dis. 2008. PMID: 18502655 Free PMC article.
-
Genetic mouse models of Huntington's disease: focus on electrophysiological mechanisms.ASN Neuro. 2010 Apr 7;2(2):e00033. doi: 10.1042/AN20090058. ASN Neuro. 2010. PMID: 20396376 Free PMC article. Review.
-
Functional Differences Between Direct and Indirect Striatal Output Pathways in Huntington's Disease.J Huntingtons Dis. 2012;1(1):17-25. doi: 10.3233/JHD-2012-120009. J Huntingtons Dis. 2012. PMID: 25063187 Free PMC article. Review.
Cited by
-
In vivo Dopamine Efflux is Decreased in Striatum of both Fragment (R6/2) and Full-Length (YAC128) Transgenic Mouse Models of Huntington's Disease.Front Syst Neurosci. 2011 Jul 15;5:61. doi: 10.3389/fnsys.2011.00061. eCollection 2011. Front Syst Neurosci. 2011. PMID: 21811446 Free PMC article.
-
Cortical and Striatal Circuits in Huntington's Disease.Front Neurosci. 2020 Feb 6;14:82. doi: 10.3389/fnins.2020.00082. eCollection 2020. Front Neurosci. 2020. PMID: 32116525 Free PMC article. Review.
-
Disrupted striatal neuron inputs and outputs in Huntington's disease.CNS Neurosci Ther. 2018 Apr;24(4):250-280. doi: 10.1111/cns.12844. CNS Neurosci Ther. 2018. PMID: 29582587 Free PMC article. Review.
-
Huntington's disease mouse models: unraveling the pathology caused by CAG repeat expansion.Fac Rev. 2021 Oct 21;10:77. doi: 10.12703/r/10-77. eCollection 2021. Fac Rev. 2021. PMID: 34746930 Free PMC article. Review.
-
Dysregulation of protein SUMOylation networks in Huntington's disease R6/2 mouse striatum.Brain. 2025 Apr 3;148(4):1212-1227. doi: 10.1093/brain/awae319. Brain. 2025. PMID: 39391934 Free PMC article.
References
-
- Bates G, Harper PS, Jones L. Huntington's Disease, 3rd edn, Oxford University Press, Oxford and New York. 2002
-
- Bates GP, Hockly E. Experimental therapeutics in Huntington's disease: are models useful for therapeutic trials? Curr Opin Neurol. 2003;16:465–470. - PubMed
-
- Cepeda C, Ariano MA, Calvert CR, Flores-Hernandez J, Chandler SH, Leavitt BR, Hayden MR, Levine MS. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res. 2001;66:525–539. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
