

Novel deletion alleles carrying CYP21A1P/A2 chimeric genes in Brazilian patients with 21-hydroxylase deficiency
- PMID: 20587039
- PMCID: PMC3161346
- DOI: 10.1186/1471-2350-11-104
Novel deletion alleles carrying CYP21A1P/A2 chimeric genes in Brazilian patients with 21-hydroxylase deficiency
Abstract
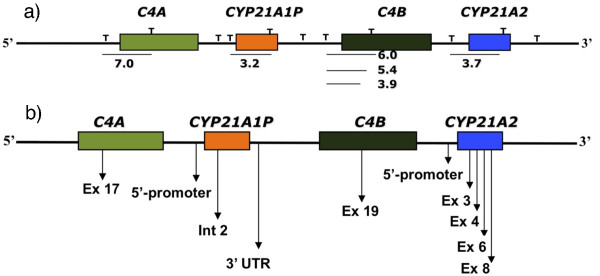
Background: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency is caused by deletions, large gene conversions or mutations in CYP21A2 gene. The human gene is located at 6p21.3 within a locus containing the genes for putative serine/threonine Kinase RP, complement C4, steroid 21-hydroxylase CYP21 tenascin TNX, normally, in a duplicated cluster known as RCCX module. The CYP21 extra copy is a pseudogene (CYP21A1P). In Brazil, 30-kb deletion forming monomodular alleles that carry chimeric CYP21A1P/A2 genes corresponds to ~9% of disease-causing alleles. Such alleles are considered to result from unequal crossovers within the bimodular C4/CYP21 locus. Depending on the localization of recombination breakpoint, different alleles can be generated conferring the locus high degree of allelic variability. The purpose of the study was to investigate the variability of deleted alleles in patients with 21-hydroxylase deficiency.
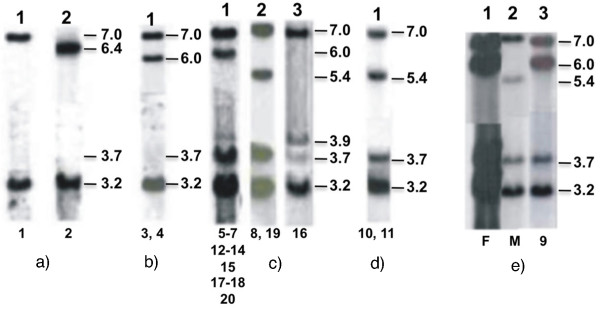
Methods: We used different techniques to investigate the variability of 30-kb deletion alleles in patients with 21-hydroxylase deficiency. Alleles were first selected after Southern blotting. The composition of CYP21A1P/A2 chimeric genes was investigated by ASO-PCR and MLPA analyses followed by sequencing to refine the location of recombination breakpoints. Twenty patients carrying at least one allele with C4/CYP21 30-kb deletion were included in the study.
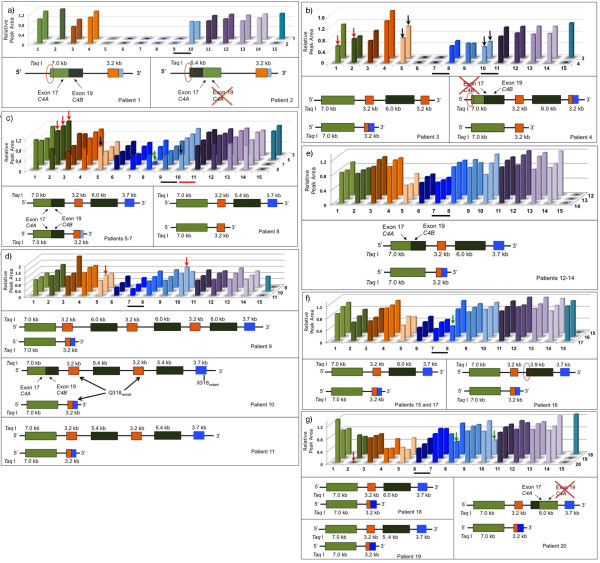
Results: An allele carrying a CYP21A1P/A2 chimeric gene was found unusually associated to a C4B/C4A Taq I 6.4-kb fragment, generally associated to C4B and CYP21A1P deletions. A novel haplotype bearing both p.P34L and p.H62L, novel and rare mutations, respectively, was identified in exon 1, however p.P30L, the most frequent pseudogene-derived mutation in this exon, was absent. Four unrelated patients showed this haplotype. Absence of p.P34L in CYP21A1P of normal controls indicated that it is not derived from pseudogene. In addition, the combination of different approaches revealed nine haplotypes for deleted 21-hydroxylase deficiency alleles.
Conclusions: This study demonstrated high allelic variability for 30-kb deletion in patients with 21-hydroxylase deficiency indicating that a founder effect might be improbable for most monomodular alleles carrying CYP21A1P/A2 chimeric genes in Brazil.
Figures
Similar articles
-
A rational, non-radioactive strategy for the molecular diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency.Gene. 2013 Sep 10;526(2):239-45. doi: 10.1016/j.gene.2013.03.082. Epub 2013 Apr 6. Gene. 2013. PMID: 23570880
-
A new CYP21A1P/CYP21A2 chimeric gene identified in an Italian woman suffering from classical congenital adrenal hyperplasia form.BMC Med Genet. 2009 Jul 22;10:72. doi: 10.1186/1471-2350-10-72. BMC Med Genet. 2009. PMID: 19624807 Free PMC article.
-
Chimeric CYP21A1P/CYP21A2 genes identified in Czech patients with congenital adrenal hyperplasia.Eur J Med Genet. 2011 Mar-Apr;54(2):112-7. doi: 10.1016/j.ejmg.2010.10.005. Epub 2010 Oct 21. Eur J Med Genet. 2011. PMID: 20970527
-
Genes and Pseudogenes: Complexity of the RCCX Locus and Disease.Front Endocrinol (Lausanne). 2021 Jul 30;12:709758. doi: 10.3389/fendo.2021.709758. eCollection 2021. Front Endocrinol (Lausanne). 2021. PMID: 34394006 Free PMC article. Review.
-
Variants of the CYP21A2 and CYP21A1P genes in congenital adrenal hyperplasia.Clin Chim Acta. 2013 Mar 15;418:37-44. doi: 10.1016/j.cca.2012.12.030. Epub 2013 Jan 9. Clin Chim Acta. 2013. PMID: 23313747 Review.
Cited by
-
Mutational characterization of congenital adrenal hyperplasia due to 21-hydroxylase deficiency in Malaysia.J Endocrinol Invest. 2013 Jun;36(6):366-74. doi: 10.3275/8648. Epub 2012 Oct 1. J Endocrinol Invest. 2013. PMID: 23027774
-
Junction site analysis of chimeric CYP21A1P/CYP21A2 genes in 21-hydroxylase deficiency.Clin Chem. 2012 Feb;58(2):421-30. doi: 10.1373/clinchem.2011.174037. Epub 2011 Dec 7. Clin Chem. 2012. PMID: 22156666 Free PMC article.
-
Identification of a novel compound heterozygous mutation of the CYP21A2 gene causing 21‑hydroxylase deficiency in a Chinese pedigree.Mol Med Rep. 2018 Mar;17(3):4265-4272. doi: 10.3892/mmr.2018.8391. Epub 2018 Jan 8. Mol Med Rep. 2018. PMID: 29328376 Free PMC article.
-
Common disease-associated gene variants in a Saudi Arabian population.Ann Saudi Med. 2022 Jan-Feb;42(1):29-35. doi: 10.5144/0256-4947.2022.29. Epub 2022 Feb 3. Ann Saudi Med. 2022. PMID: 35112591 Free PMC article.
-
Characterization of Mutations Causing CYP21A2 Deficiency in Brazilian and Portuguese Populations.Int J Mol Sci. 2021 Dec 28;23(1):296. doi: 10.3390/ijms23010296. Int J Mol Sci. 2021. PMID: 35008721 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
