

Co-regulation of nuclear respiratory factor-1 by NFkappaB and CREB links LPS-induced inflammation to mitochondrial biogenesis
- PMID: 20587593
- PMCID: PMC2912462
- DOI: 10.1242/jcs.064089
Co-regulation of nuclear respiratory factor-1 by NFkappaB and CREB links LPS-induced inflammation to mitochondrial biogenesis
Abstract
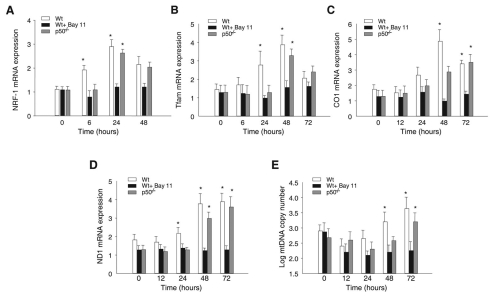
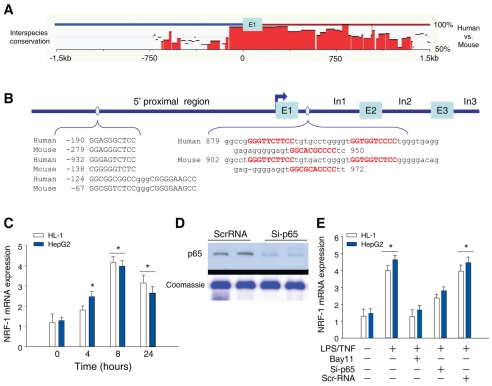
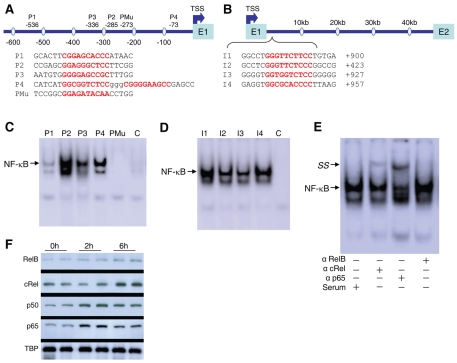
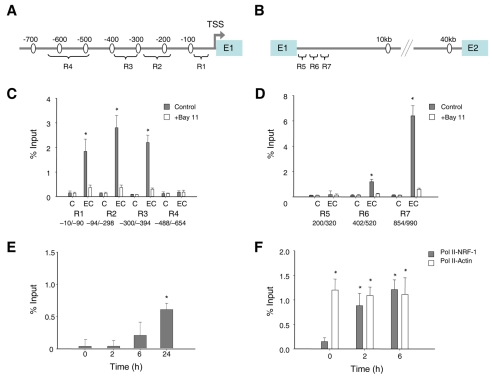
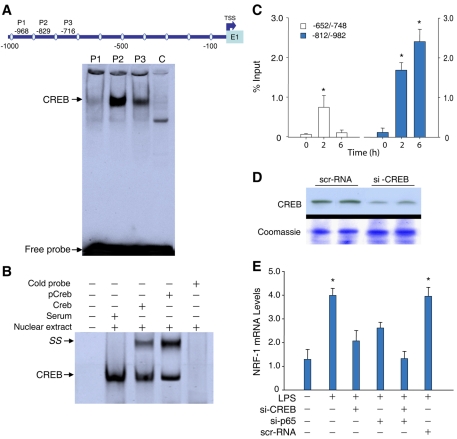
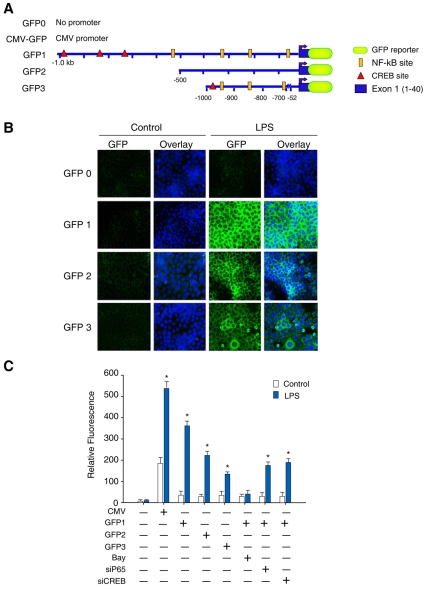
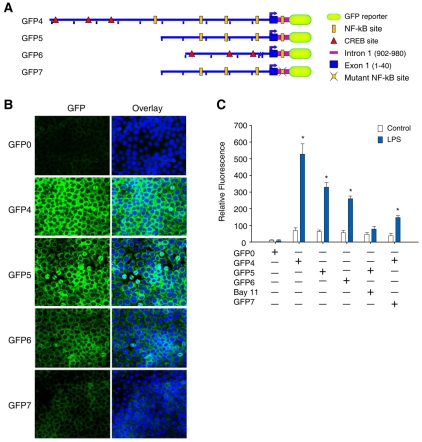
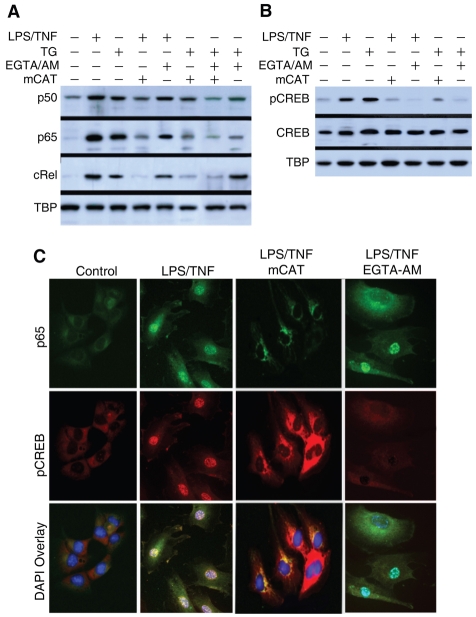
The nuclear respiratory factor-1 (NRF1) gene is activated by lipopolysaccharide (LPS), which might reflect TLR4-mediated mitigation of cellular inflammatory damage via initiation of mitochondrial biogenesis. To test this hypothesis, we examined NRF1 promoter regulation by NFκB, and identified interspecies-conserved κB-responsive promoter and intronic elements in the NRF1 locus. In mice, activation of Nrf1 and its downstream target, Tfam, by Escherichia coli was contingent on NFκB, and in LPS-treated hepatocytes, NFκB served as an NRF1 enhancer element in conjunction with NFκB promoter binding. Unexpectedly, optimal NRF1 promoter activity after LPS also required binding by the energy-state-dependent transcription factor CREB. EMSA and ChIP assays confirmed p65 and CREB binding to the NRF1 promoter and p65 binding to intron 1. Functionality for both transcription factors was validated by gene-knockdown studies. LPS regulation of NRF1 led to mtDNA-encoded gene expression and expansion of mtDNA copy number. In cells expressing plasmid constructs containing the NRF-1 promoter and GFP, LPS-dependent reporter activity was abolished by cis-acting κB-element mutations, and nuclear accumulation of NFκB and CREB demonstrated dependence on mitochondrial H(2)O(2). These findings indicate that TLR4-dependent NFκB and CREB activation co-regulate the NRF1 promoter with NFκB intronic enhancement and redox-regulated nuclear translocation, leading to downstream target-gene expression, and identify NRF-1 as an early-phase component of the host antibacterial defenses.
Figures
Similar articles
-
[Nuclear respiratory factor 1 mediates LPS-induced acute lung injury through NF-κB].Sheng Li Xue Bao. 2022 Jun 25;74(3):401-410. Sheng Li Xue Bao. 2022. PMID: 35770638 Chinese.
-
Exchange of a nuclear corepressor between NF-kappaB and CREB mediates inhibition of phosphoenolpyruvate carboxykinase transcription by NF-kappaB.Chin Med J (Engl). 2010 Jan 20;123(2):221-6. Chin Med J (Engl). 2010. PMID: 20137375
-
Coordinated transcriptional regulation of Hspa1a gene by multiple transcription factors: crucial roles for HSF-1, NF-Y, NF-κB, and CREB.J Mol Biol. 2014 Jan 9;426(1):116-35. doi: 10.1016/j.jmb.2013.09.008. Epub 2013 Sep 14. J Mol Biol. 2014. PMID: 24041570
-
Nuclear respiratory factor 1 controls myocyte enhancer factor 2A transcription to provide a mechanism for coordinate expression of respiratory chain subunits.J Biol Chem. 2008 May 2;283(18):11935-46. doi: 10.1074/jbc.M707389200. Epub 2008 Jan 24. J Biol Chem. 2008. PMID: 18222924 Free PMC article.
-
Molecular basis of the association between transcription regulators nuclear respiratory factor 1 and inhibitor of DNA binding protein 3 and the development of microvascular lesions.Microvasc Res. 2022 May;141:104337. doi: 10.1016/j.mvr.2022.104337. Epub 2022 Feb 7. Microvasc Res. 2022. PMID: 35143811 Free PMC article. Review.
Cited by
-
Exogenous Alpha-Synuclein Evoked Parkin Downregulation Promotes Mitochondrial Dysfunction in Neuronal Cells. Implications for Parkinson's Disease Pathology.Front Aging Neurosci. 2021 Feb 24;13:591475. doi: 10.3389/fnagi.2021.591475. eCollection 2021. Front Aging Neurosci. 2021. PMID: 33716707 Free PMC article.
-
Mitochondrial biogenesis contributes to ischemic neuroprotection afforded by LPS pre-conditioning.J Neurochem. 2012 Nov;123 Suppl 2(0 2):125-37. doi: 10.1111/j.1471-4159.2012.07951.x. J Neurochem. 2012. PMID: 23050650 Free PMC article.
-
miR‑146a improves hepatic lipid and glucose metabolism by targeting MED1.Int J Mol Med. 2020 Feb;45(2):543-555. doi: 10.3892/ijmm.2019.4443. Epub 2019 Dec 27. Int J Mol Med. 2020. PMID: 31894315 Free PMC article.
-
DHEA metabolites activate estrogen receptors alpha and beta.Steroids. 2013 Jan;78(1):15-25. doi: 10.1016/j.steroids.2012.10.002. Epub 2012 Nov 2. Steroids. 2013. PMID: 23123738 Free PMC article.
-
Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis.Am J Respir Crit Care Med. 2012 Apr 15;185(8):851-61. doi: 10.1164/rccm.201106-1152OC. Epub 2012 Feb 3. Am J Respir Crit Care Med. 2012. PMID: 22312014 Free PMC article.
References
-
- Bakkar N., Wang J., Ladner K. J., Wang H., Dahlman J. M., Carathers M., Acharyya S., Rudnicki M. A., Hollenbach A. D., Guttridge D. C. (2008). IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J. Cell Biol. 180, 787-802 - PMC - PubMed
-
- Bergeron R., Ren J. M., Cadman K. S., Moore I. K., Perret P., Pypaert M., Young L. H., Semenkovich C. F., Shulman G. I. (2001). Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 281, E1340-E1346 - PubMed
-
- Charital Y. M., van Haasteren G., Massiha A., Schlegel W., Fujita T. (2009). A functional NF-kappaB enhancer element in the first intron contributes to the control of c-fos transcription. Gene 430, 116-122 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
