

RNA-seq analysis of sulfur-deprived Chlamydomonas cells reveals aspects of acclimation critical for cell survival
- PMID: 20587772
- PMCID: PMC2910963
- DOI: 10.1105/tpc.109.071167
RNA-seq analysis of sulfur-deprived Chlamydomonas cells reveals aspects of acclimation critical for cell survival
Erratum in
- Plant Cell. 2011 Apr;23(4):1679-81
Abstract
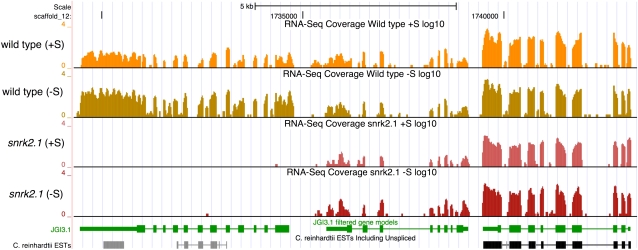
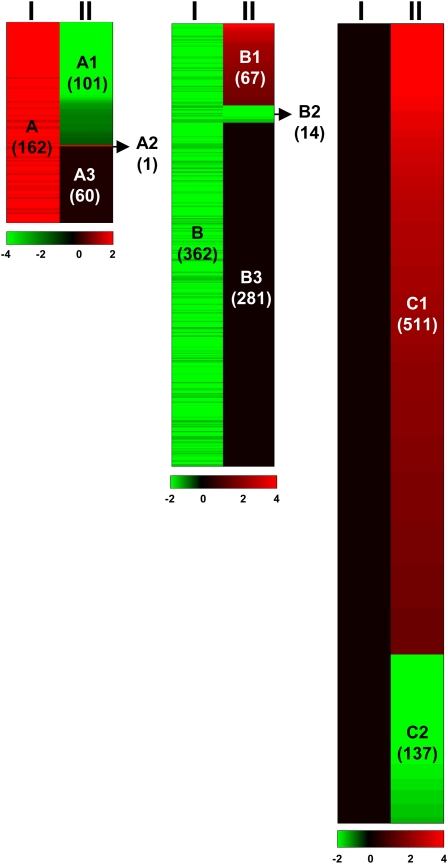
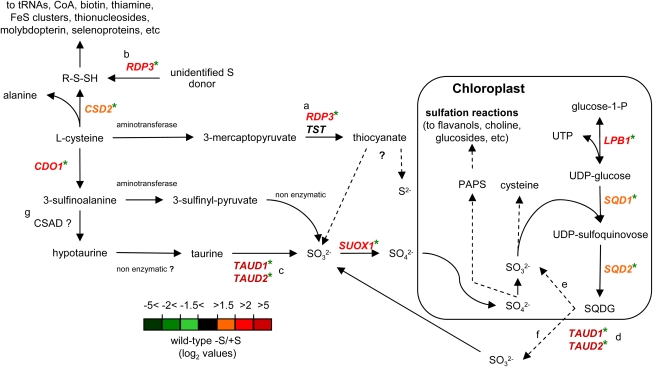
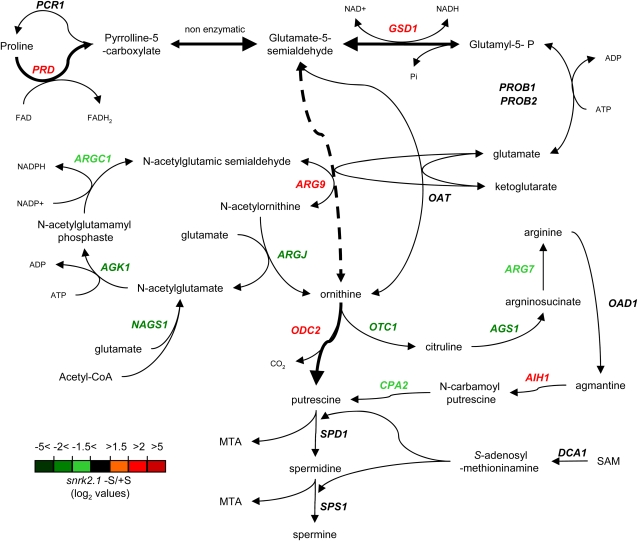
The Chlamydomonas reinhardtii transcriptome was characterized from nutrient-replete and sulfur-depleted wild-type and snrk2.1 mutant cells. This mutant is null for the regulatory Ser-Thr kinase SNRK2.1, which is required for acclimation of the alga to sulfur deprivation. The transcriptome analyses used microarray hybridization and RNA-seq technology. Quantitative RT-PCR evaluation of the results obtained by these techniques showed that RNA-seq reports a larger dynamic range of expression levels than do microarray hybridizations. Transcripts responsive to sulfur deprivation included those encoding proteins involved in sulfur acquisition and assimilation, synthesis of sulfur-containing metabolites, Cys degradation, and sulfur recycling. Furthermore, we noted potential modifications of cellular structures during sulfur deprivation, including the cell wall and complexes associated with the photosynthetic apparatus. Moreover, the data suggest that sulfur-deprived cells accumulate proteins with fewer sulfur-containing amino acids. Most of the sulfur deprivation responses are controlled by the SNRK2.1 protein kinase. The snrk2.1 mutant exhibits a set of unique responses during both sulfur-replete and sulfur-depleted conditions that are not observed in wild-type cells; the inability of this mutant to acclimate to S deprivation probably leads to elevated levels of singlet oxygen and severe oxidative stress, which ultimately causes cell death. The transcriptome results for wild-type and mutant cells strongly suggest the occurrence of massive changes in cellular physiology and metabolism as cells become depleted for sulfur and reveal aspects of acclimation that are likely critical for cell survival.
Figures







References
-
- Apel K., Hirt H. (2004). Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 55: 373–399 - PubMed
-
- Audic S., Claverie J.M. (1997). The significance of digital gene expression profiles. Genome Res. 7: 986–995 - PubMed
-
- Bentley R., Chasteen T.G. (2004). Environmental VOSCs-formation and degradation of dimethyl sulfide, methanethiol and related materials. Chemosphere 55: 291–317 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
