

RhoA and DIAPH1 mediate adrenocorticotropin-stimulated cortisol biosynthesis by regulating mitochondrial trafficking
- PMID: 20591975
- PMCID: PMC2940507
- DOI: 10.1210/en.2010-0044
RhoA and DIAPH1 mediate adrenocorticotropin-stimulated cortisol biosynthesis by regulating mitochondrial trafficking
Abstract
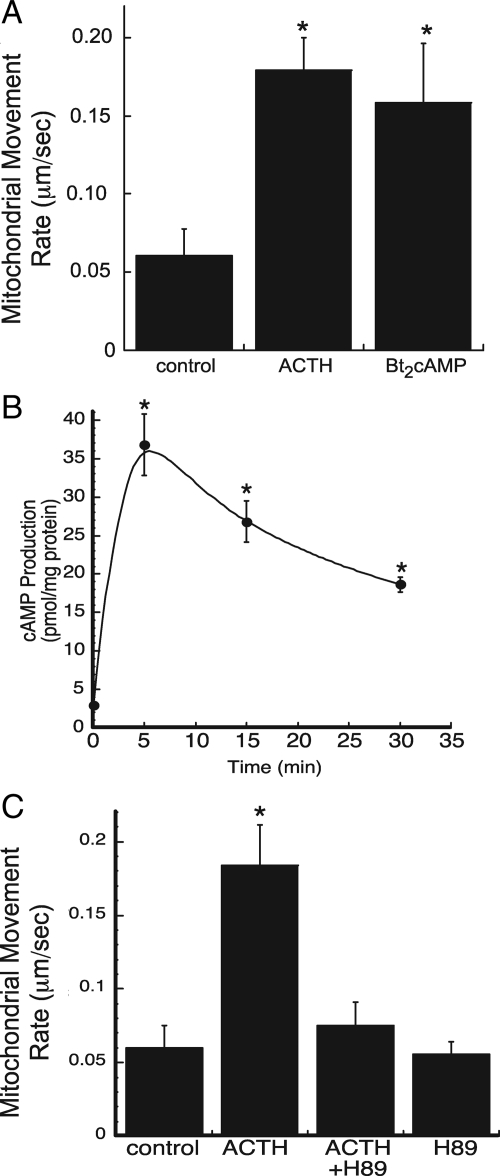
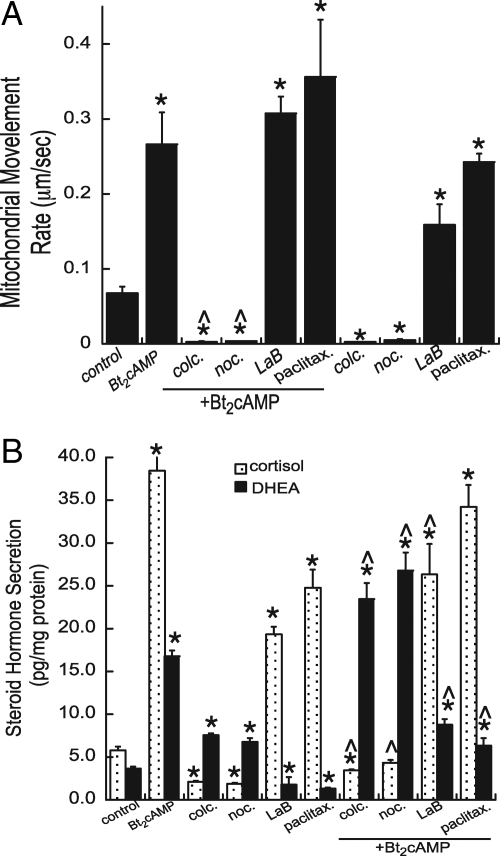
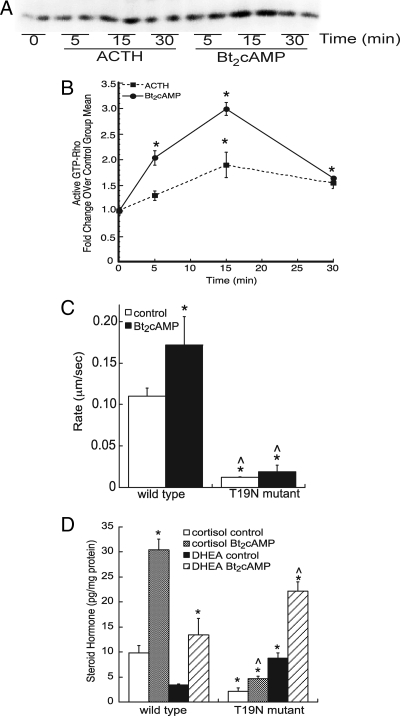
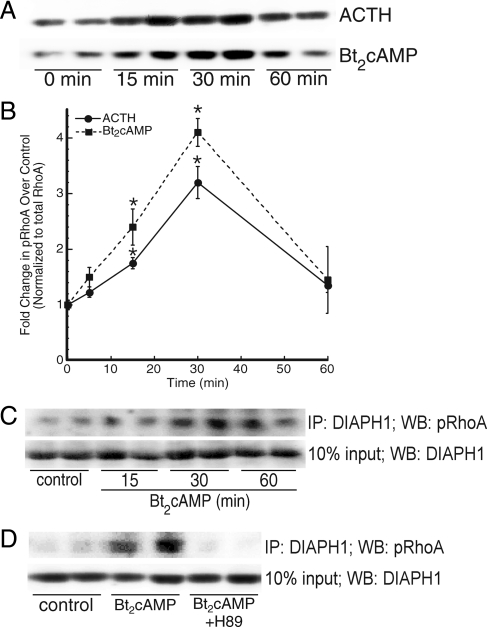
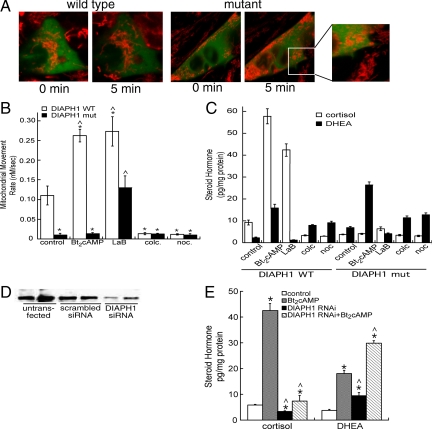
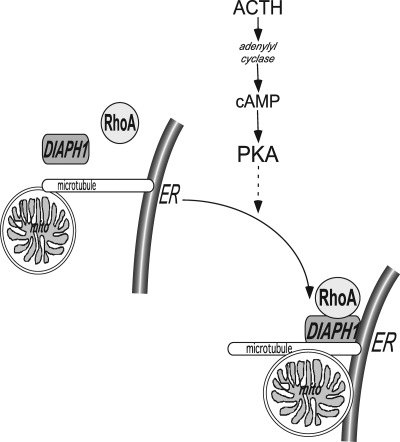
Steroid hormones are formed by the successive action of enzymes that are localized in mitochondria and the endoplasmic reticulum (ER). Compartmentalization of these enzymes in different subcellular organelles dictates the need for efficient transfer of intermediary metabolites between the mitochondrion and ER; however, the molecular determinants that regulate interorganelle substrate exchange are unknown. The objective of this study was to define the molecular mechanism by which adrenocorticotropin (ACTH) signaling regulates communication between mitochondria and the ER during steroidogenesis. Using live cell video confocal microscopy, we found that ACTH and dibutyryl cAMP rapidly increased the rate of mitochondrial movement. Inhibiting tubulin polymerization prevented both basal and ACTH/cAMP-stimulated mitochondrial trafficking and decreased cortisol secretion. This decrease in cortisol secretion evoked by microtubule inhibition was paralleled by an increase in dehydroepiandrosterone production. In contrast, treatment with paclitaxel to stabilize microtubules or latrunculin B to inhibit actin polymerization and disrupt microfilament organization increased both mitochondrial trafficking and cortisol biosynthesis. ACTH-stimulated mitochondrial movement was dependent on RhoA and the RhoA effector, diaphanous-related homolog 1 (DIAPH1). ACTH signaling temporally increased the cellular concentrations of GTP-bound and Ser-188 phosphorylated RhoA, which promoted interaction with DIAPH1. Expression of a dominant-negative RhoA mutant or silencing DIAPH1 impaired mitochondrial trafficking and cortisol biosynthesis and concomitantly increased the secretion of adrenal androgens. We conclude that ACTH regulates cortisol production by facilitating interorganelle substrate transfer via a process that is mediated by RhoA and DIAPH1, which act to coordinate the dynamic trafficking of mitochondria.
Figures
Similar articles
-
cAMP-stimulated phosphorylation of diaphanous 1 regulates protein stability and interaction with binding partners in adrenocortical cells.Mol Biol Cell. 2013 Mar;24(6):848-57. doi: 10.1091/mbc.E12-08-0597. Epub 2013 Jan 16. Mol Biol Cell. 2013. PMID: 23325789 Free PMC article.
-
Regulation of adrenocortical steroid hormone production by RhoA-diaphanous 1 signaling and the cytoskeleton.Mol Cell Endocrinol. 2013 May 22;371(1-2):79-86. doi: 10.1016/j.mce.2012.11.014. Epub 2012 Nov 24. Mol Cell Endocrinol. 2013. PMID: 23186810 Free PMC article. Review.
-
The scaffold-protein IQGAP1 enhances and spatially restricts the actin-nucleating activity of Diaphanous-related formin 1 (DIAPH1).J Biol Chem. 2020 Mar 6;295(10):3134-3147. doi: 10.1074/jbc.RA119.010476. Epub 2020 Jan 31. J Biol Chem. 2020. PMID: 32005666 Free PMC article.
-
Negative cooperativity regulates ligand activation of DIAPH1 and other diaphanous related formins.Commun Biol. 2025 May 21;8(1):776. doi: 10.1038/s42003-025-08222-5. Commun Biol. 2025. PMID: 40399622 Free PMC article.
-
Regulation of steroid hormone biosynthesis by the cytoskeleton.Lipids. 2008 Dec;43(12):1109-15. doi: 10.1007/s11745-008-3221-2. Epub 2008 Aug 26. Lipids. 2008. PMID: 18726632 Free PMC article. Review.
Cited by
-
Unprenylated RhoA contributes to IL-1β hypersecretion in mevalonate kinase deficiency model through stimulation of Rac1 activity.J Biol Chem. 2014 Oct 3;289(40):27757-65. doi: 10.1074/jbc.M114.571810. Epub 2014 Aug 8. J Biol Chem. 2014. PMID: 25107911 Free PMC article.
-
Plasma Diaphanous Related Formin 1 Levels Are Associated with Altered Glucose Metabolism and Insulin Resistance in Patients with Polycystic Ovary Syndrome: A Case Control Study.Mediators Inflamm. 2022 Feb 11;2022:9620423. doi: 10.1155/2022/9620423. eCollection 2022. Mediators Inflamm. 2022. PMID: 35185386 Free PMC article.
-
cAMP-stimulated phosphorylation of diaphanous 1 regulates protein stability and interaction with binding partners in adrenocortical cells.Mol Biol Cell. 2013 Mar;24(6):848-57. doi: 10.1091/mbc.E12-08-0597. Epub 2013 Jan 16. Mol Biol Cell. 2013. PMID: 23325789 Free PMC article.
-
Role of Protein Phosphorylation and Tyrosine Phosphatases in the Adrenal Regulation of Steroid Synthesis and Mitochondrial Function.Front Endocrinol (Lausanne). 2016 Jun 9;7:60. doi: 10.3389/fendo.2016.00060. eCollection 2016. Front Endocrinol (Lausanne). 2016. PMID: 27375556 Free PMC article. Review.
-
Rab40-Cullin5 complex regulates EPLIN and actin cytoskeleton dynamics during cell migration.J Cell Biol. 2021 Jul 5;220(7):e202008060. doi: 10.1083/jcb.202008060. Epub 2021 May 17. J Cell Biol. 2021. PMID: 33999101 Free PMC article.
References
-
- Arlt W, Stewart PM 2005 Adrenal corticosteroid biosynthesis, metabolism, and action. Endocrinol Metab Clin North Am 34:293–313, viii - PubMed
-
- Miller WL 2002 Androgen biosynthesis from cholesterol to DHEA. Mol Cell Endocrinol 198:7–14 - PubMed
-
- Waterman MR, Keeney DS 1996 Diverse molecular mechanisms regulate the expression of steroid hydroxylase genes required for production of ligands for nuclear receptors. In: Jefcoate CR, ed. Physiological functions of cytochrome P450 in relation to structure and regulation. Greenwich, CT: JAI Press, Inc.; 81–102
-
- Sewer MB, Waterman MR 2003 ACTH modulation of transcription factors responsible for steroid hydroxylase gene expression in the adrenal cortex. Microsc Res Tech 61:300–307 - PubMed
-
- Sewer MB, Dammer EB, Jagarlapudi S 2007 Transcriptional regulation of adrenocortical steroidogenic gene expression. Drug Metab Rev 39:371–388 - PubMed
