

Analysis of spliceosomal proteins in Trypanosomatids reveals novel functions in mRNA processing
- PMID: 20592024
- PMCID: PMC2934664
- DOI: 10.1074/jbc.M109.095349
Analysis of spliceosomal proteins in Trypanosomatids reveals novel functions in mRNA processing
Abstract
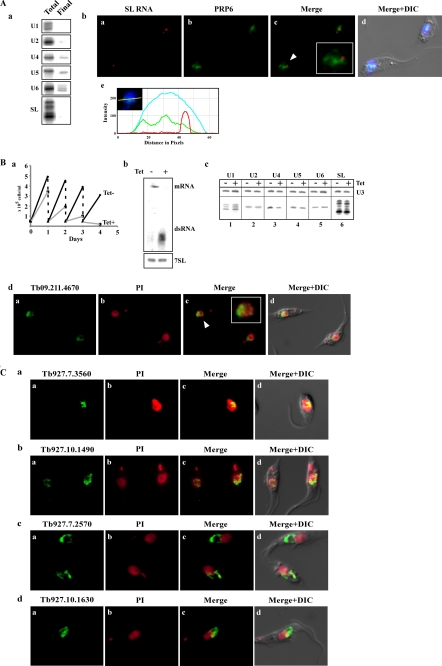
In trypanosomatids, all mRNAs are processed via trans-splicing, although cis-splicing also occurs. In trans-splicing, a common small exon, the spliced leader (SL), which is derived from a small SL RNA species, is added to all mRNAs. Sm and Lsm proteins are core proteins that bind to U snRNAs and are essential for both these splicing processes. In this study, SmD3- and Lsm3-associated complexes were purified to homogeneity from Leishmania tarentolae. The purified complexes were analyzed by mass spectrometry, and 54 and 39 proteins were purified from SmD3 and Lsm complexes, respectively. Interestingly, among the proteins purified from Lsm3, no mRNA degradation factors were detected, as in Lsm complexes from other eukaryotes. The U1A complex was purified and mass spectrometry analysis identified, in addition to U1 small nuclear ribonucleoprotein (snRNP) proteins, additional co-purified proteins, including the polyadenylation factor CPSF73. Defects observed in cells silenced for U1 snRNP proteins suggest that the U1 snRNP functions exclusively in cis-splicing, although U1A also participates in polyadenylation and affects trans-splicing. The study characterized several trypanosome-specific nuclear factors involved in snRNP biogenesis, whose function was elucidated in Trypanosoma brucei. Conserved factors, such as PRP19, which functions at the heart of every cis-spliceosome, also affect SL RNA modification; GEMIN2, a protein associated with SMN (survival of motor neurons) and implicated in selective association of U snRNA with core Sm proteins in trypanosomes, is a master regulator of snRNP assembly. This study demonstrates the existence of trypanosomatid-specific splicing factors but also that conserved snRNP proteins possess trypanosome-specific functions.
Figures







Similar articles
-
Spliceosomal proteomics in Trypanosoma brucei reveal new RNA splicing factors.Eukaryot Cell. 2009 Jul;8(7):990-1000. doi: 10.1128/EC.00075-09. Epub 2009 May 8. Eukaryot Cell. 2009. PMID: 19429779 Free PMC article.
-
Direct analysis of nematode cis- and trans-spliceosomes: a functional role for U5 snRNA in spliced leader addition trans-splicing and the identification of novel Sm snRNPs.RNA. 1996 Aug;2(8):735-45. RNA. 1996. PMID: 8752084 Free PMC article.
-
Genome-wide RNA-binding analysis of the trypanosome U1 snRNP proteins U1C and U1-70K reveals cis/trans-spliceosomal network.Nucleic Acids Res. 2014 Jun;42(10):6603-15. doi: 10.1093/nar/gku286. Epub 2014 Apr 19. Nucleic Acids Res. 2014. PMID: 24748659 Free PMC article.
-
The pre-mRNA splicing machinery of trypanosomes: complex or simplified?Eukaryot Cell. 2010 Aug;9(8):1159-70. doi: 10.1128/EC.00113-10. Epub 2010 Jun 25. Eukaryot Cell. 2010. PMID: 20581293 Free PMC article. Review.
-
mRNA splicing in trypanosomes.Int J Med Microbiol. 2012 Oct;302(4-5):221-4. doi: 10.1016/j.ijmm.2012.07.004. Epub 2012 Sep 7. Int J Med Microbiol. 2012. PMID: 22964417 Review.
Cited by
-
The trypanocidal benzoxaborole AN7973 inhibits trypanosome mRNA processing.PLoS Pathog. 2018 Sep 25;14(9):e1007315. doi: 10.1371/journal.ppat.1007315. eCollection 2018 Sep. PLoS Pathog. 2018. PMID: 30252911 Free PMC article.
-
Arabidopsis thaliana LSM proteins function in mRNA splicing and degradation.Nucleic Acids Res. 2013 Jul;41(12):6232-49. doi: 10.1093/nar/gkt296. Epub 2013 Apr 24. Nucleic Acids Res. 2013. PMID: 23620288 Free PMC article.
-
Tudor staphylococcal nuclease (Tudor-SN) participates in small ribonucleoprotein (snRNP) assembly via interacting with symmetrically dimethylated Sm proteins.J Biol Chem. 2012 May 25;287(22):18130-41. doi: 10.1074/jbc.M111.311852. Epub 2012 Apr 9. J Biol Chem. 2012. PMID: 22493508 Free PMC article.
-
Structural basis of Spliced Leader RNA recognition by the Trypanosoma brucei cap-binding complex.Nat Commun. 2025 Jan 15;16(1):685. doi: 10.1038/s41467-024-55373-w. Nat Commun. 2025. PMID: 39814716 Free PMC article.
-
Regulation of gene expression in trypanosomatids: living with polycistronic transcription.Open Biol. 2019 Jun 28;9(6):190072. doi: 10.1098/rsob.190072. Epub 2019 Jun 5. Open Biol. 2019. PMID: 31164043 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
