

A principled approach to deriving approximate conditional sampling distributions in population genetics models with recombination
- PMID: 20592264
- PMCID: PMC2940296
- DOI: 10.1534/genetics.110.117986
A principled approach to deriving approximate conditional sampling distributions in population genetics models with recombination
Abstract
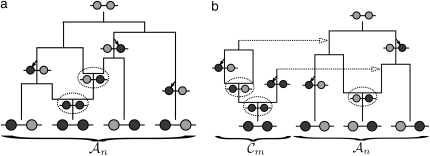
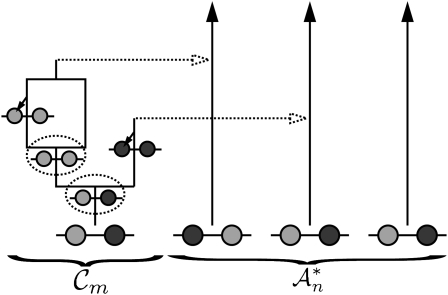
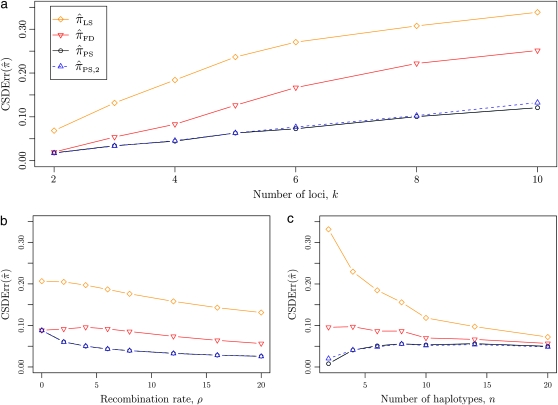
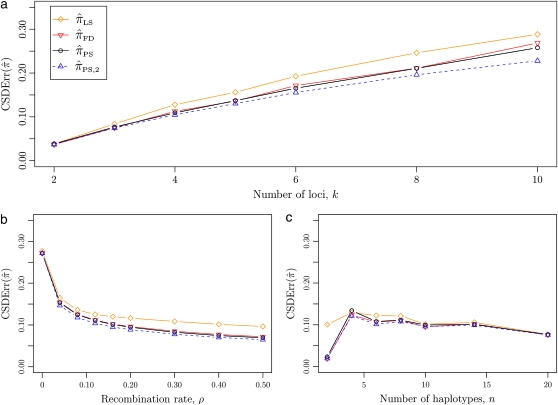
The multilocus conditional sampling distribution (CSD) describes the probability that an additionally sampled DNA sequence is of a certain type, given that a collection of sequences has already been observed. The CSD has a wide range of applications in both computational biology and population genomics analysis, including phasing genotype data into haplotype data, imputing missing data, estimating recombination rates, inferring local ancestry in admixed populations, and importance sampling of coalescent genealogies. Unfortunately, the true CSD under the coalescent with recombination is not known, so approximations, formulated as hidden Markov models, have been proposed in the past. These approximations have led to a number of useful statistical tools, but it is important to recognize that they were not derived from, though were certainly motivated by, principles underlying the coalescent process. The goal of this article is to develop a principled approach to derive improved CSDs directly from the underlying population genetics model. Our approach is based on the diffusion process approximation and the resulting mathematical expressions admit intuitive genealogical interpretations, which we utilize to introduce further approximations and make our method scalable in the number of loci. The general algorithm presented here applies to an arbitrary number of loci and an arbitrary finite-alleles recurrent mutation model. Empirical results are provided to demonstrate that our new CSDs are in general substantially more accurate than previously proposed approximations.
Figures
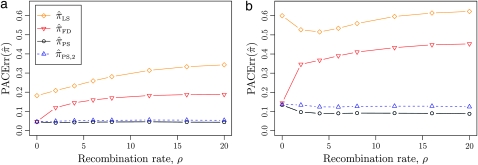
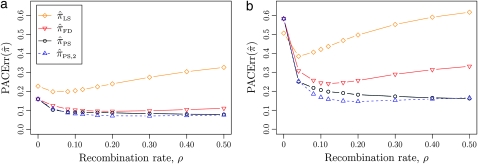
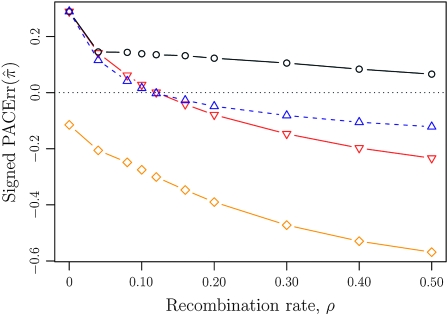
References
-
- Crawford, D. C., T. Bhangale, N. Li, G. Hellenthal, M. J. Rieder et al., 2004. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat. Genet. 36 700–706. - PubMed
-
- De Iorio, M., and R. C. Griffiths, 2004. a Importance sampling on coalescent histories I. Adv. Appl. Probab. 36 417–433.
-
- De Iorio, M., and R. C. Griffiths, 2004. b Importance sampling on coalescent histories II. Adv. Appl. Probab. 36 434–454.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
