

Chronic myeloid leukemia: mechanisms of blastic transformation
- PMID: 20592475
- PMCID: PMC2898591
- DOI: 10.1172/JCI41246
Chronic myeloid leukemia: mechanisms of blastic transformation
Abstract
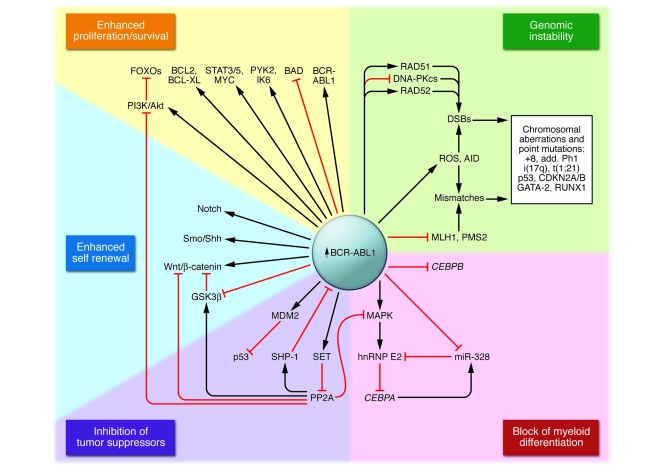
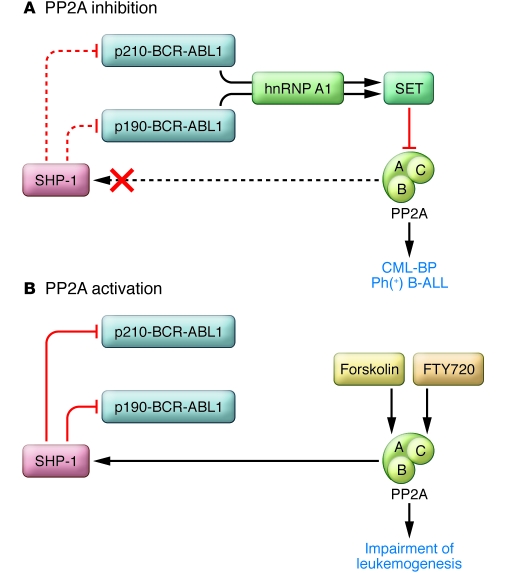
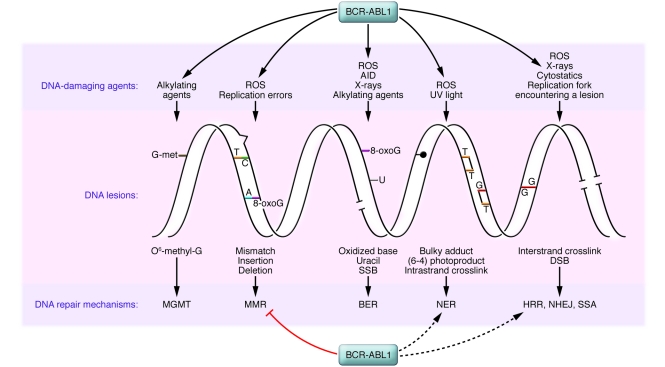
The BCR-ABL1 oncoprotein transforms pluripotent HSCs and initiates chronic myeloid leukemia (CML). Patients with early phase (also known as chronic phase [CP]) disease usually respond to treatment with ABL tyrosine kinase inhibitors (TKIs), although some patients who respond initially later become resistant. In most patients, TKIs reduce the leukemia cell load substantially, but the cells from which the leukemia cells are derived during CP (so-called leukemia stem cells [LSCs]) are intrinsically insensitive to TKIs and survive long term. LSCs or their progeny can acquire additional genetic and/or epigenetic changes that cause the leukemia to transform from CP to a more advanced phase, which has been subclassified as either accelerated phase or blastic phase disease. The latter responds poorly to treatment and is usually fatal. Here, we discuss what is known about the molecular mechanisms leading to blastic transformation of CML and propose some novel therapeutic approaches.
Figures
References
-
- Barnes DJ, Melo JV. Primitive, quiescent and difficult to kill: the role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle. 2006;5(24):2862–2866. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
