

N-methyl-aspartate receptor and neuronal nitric oxide synthase activation mediate bilirubin-induced neurotoxicity
- PMID: 20593111
- PMCID: PMC2935951
- DOI: 10.2119/molmed.2009.00152
N-methyl-aspartate receptor and neuronal nitric oxide synthase activation mediate bilirubin-induced neurotoxicity
Abstract
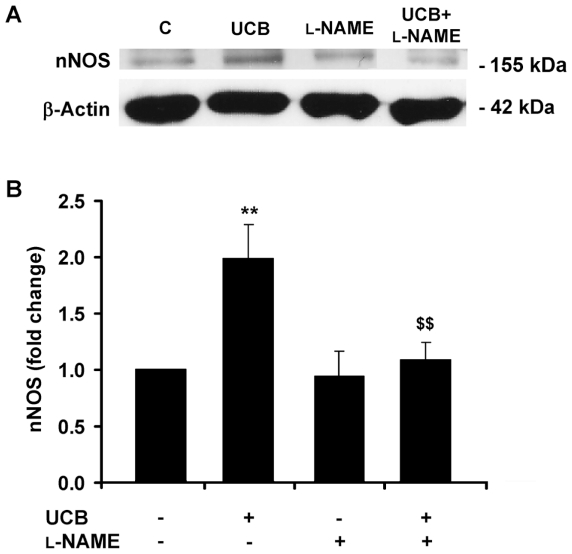
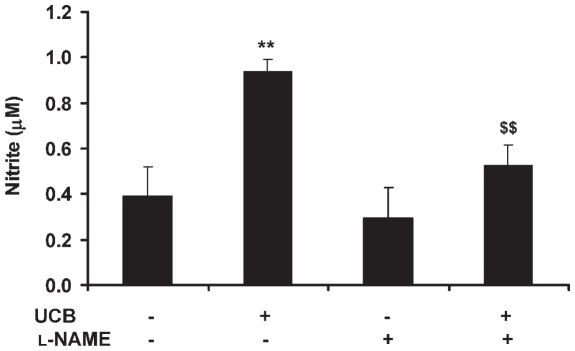
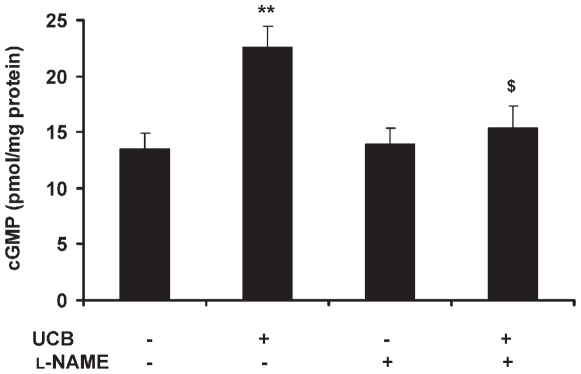
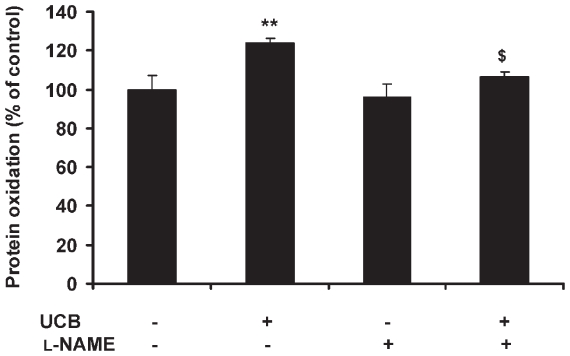
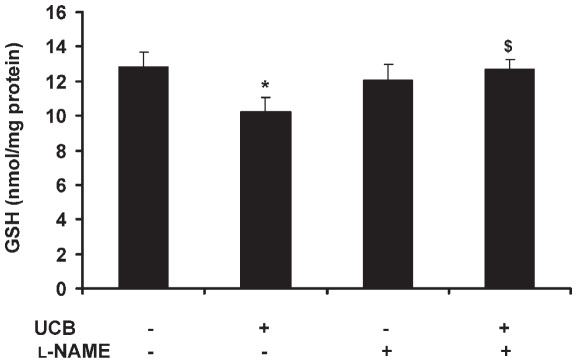
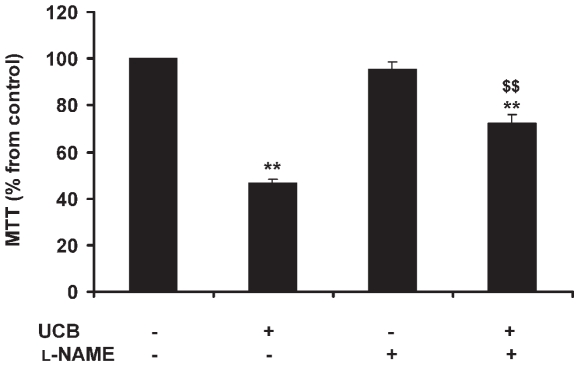
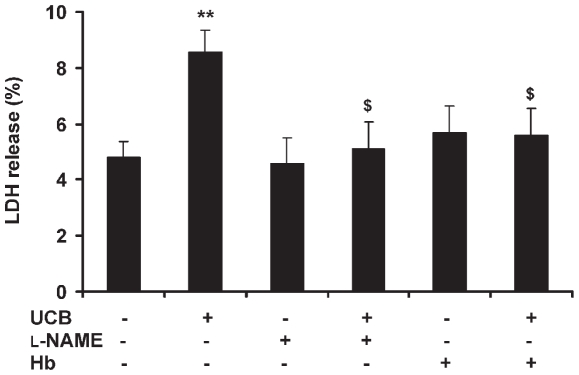
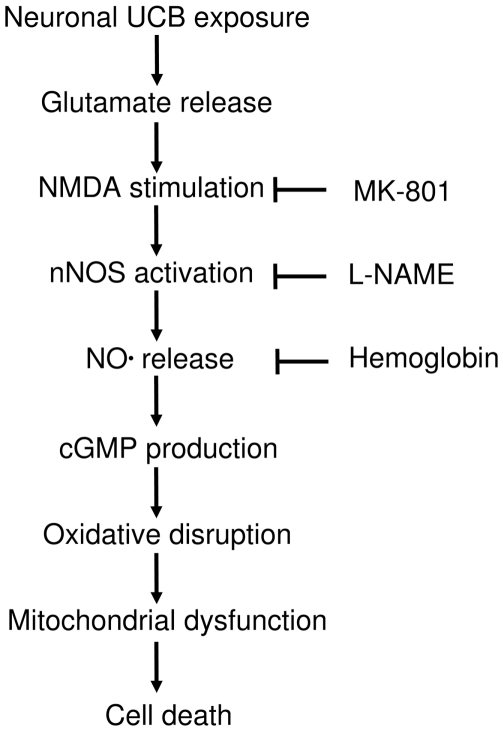
Hyperbilirubinemia may lead to neurotoxicity and neuronal death. Although the mechanisms of nerve cell damage by unconjugated bilirubin (UCB) appear to involve a disruption of the redox status and excitotoxicity, the contribution of nitric oxide (NO·) and of N-methyl-D-aspartate (NMDA) glutamate receptors is unclear. We investigated the role of NO· and NMDA glutamate receptors in the pathways of nerve cell demise by UCB. Neurons were incubated with 100 micromol/L UCB, in the presence of 100 micromol/L human serum albumin for 4 h at 37ºC, alone or in combination with N-ω-nitro-L-arginine methyl ester (L-NAME) (an inhibitor of neuronal nitric oxide synthase [nNOS]), hemoglobin (an NO· scavenger) or (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801) (an NMDA-receptor antagonist). Exposure to UCB led to increased expression of nNOS and production of both NO· and cyclic guanosine 3',5'-monophosphate (cGMP), along with protein oxidation and depletion of glutathione. These events concurred for cell dysfunction and death and were counteracted by L-NAME. Moreover, the UCB-induced loss of neuronal viability was abolished by hemoglobin, whereas the activation of nNOS and production of both NO· and cGMP were counteracted by MK-801, resulting in significant protection from cell dysfunction and death. These results reinforce the involvement of oxidative stress by showing that nerve cell damage by UCB is mediated by NO· and therefore is counteracted by NO· inhibitors or scavengers. Our findings strongly suggest that the activation of nNOS and neurotoxicity occur through the engagement of NMDA receptors. These data reveal a role for overstimulation of glutamate receptors in mediating oxidative damage by UCB.
Figures
Similar articles
-
Targeted disruption of PSD-93 gene reduces platelet-activating factor-induced neurotoxicity in cultured cortical neurons.Exp Neurol. 2004 Sep;189(1):16-24. doi: 10.1016/j.expneurol.2004.05.013. Exp Neurol. 2004. PMID: 15296832
-
Bilirubin injury to neurons: contribution of oxidative stress and rescue by glycoursodeoxycholic acid.Neurotoxicology. 2008 Mar;29(2):259-69. doi: 10.1016/j.neuro.2007.11.002. Epub 2007 Nov 19. Neurotoxicology. 2008. PMID: 18164405
-
Neuronal nitric oxide synthase and N-methyl-D-aspartate neurons in experimental carbon monoxide poisoning.Toxicol Appl Pharmacol. 2004 Feb 1;194(3):280-95. doi: 10.1016/j.taap.2003.09.017. Toxicol Appl Pharmacol. 2004. PMID: 14761684
-
Redox modulation of the NMDA receptor.Cell Mol Life Sci. 2000 Oct;57(11):1535-41. doi: 10.1007/pl00000638. Cell Mol Life Sci. 2000. PMID: 11092448 Free PMC article. Review.
-
N-methyl-d-aspartate receptors: Structure, function, and role in organophosphorus compound poisoning.Biofactors. 2024 Sep-Oct;50(5):868-884. doi: 10.1002/biof.2048. Epub 2024 Feb 28. Biofactors. 2024. PMID: 38415801 Review.
Cited by
-
Exposure to lipopolysaccharide and/or unconjugated bilirubin impair the integrity and function of brain microvascular endothelial cells.PLoS One. 2012;7(5):e35919. doi: 10.1371/journal.pone.0035919. Epub 2012 May 7. PLoS One. 2012. PMID: 22586454 Free PMC article.
-
Predictive value of different bilirubin subtypes for clinical outcomes in patients with acute ischemic stroke receiving thrombolysis therapy.CNS Neurosci Ther. 2022 Feb;28(2):226-236. doi: 10.1111/cns.13759. Epub 2021 Nov 14. CNS Neurosci Ther. 2022. PMID: 34779141 Free PMC article.
-
Factors Associated With Age-related Hearing Impairment: A Retrospective Cohort Study.Medicine (Baltimore). 2015 Oct;94(43):e1846. doi: 10.1097/MD.0000000000001846. Medicine (Baltimore). 2015. PMID: 26512592 Free PMC article.
-
Suppressed microRNA-96 inhibits iNOS expression and dopaminergic neuron apoptosis through inactivating the MAPK signaling pathway by targeting CACNG5 in mice with Parkinson's disease.Mol Med. 2018 Nov 28;24(1):61. doi: 10.1186/s10020-018-0059-9. Mol Med. 2018. PMID: 30486773 Free PMC article.
-
Lipid peroxidation, DNA damage and total antioxidant status in neonatal hyperbilirubinemia.J Perinatol. 2014 Jul;34(7):519-23. doi: 10.1038/jp.2014.45. Epub 2014 Mar 27. J Perinatol. 2014. PMID: 24674982
References
-
- Sies H. Oxidative stress: oxidants and antioxidants. Exp. Physiol. 1997;82:291–5. - PubMed
-
- Ledo A, Frade J, Barbosa RM, Laranjinha J. Nitric oxide in brain: diffusion, targets and concentration dynamics in hippocampal subregions. Mol. Aspects Med. 2004;25:75–89. - PubMed
-
- Moncada S, Bolaños JP. Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 2006;97:1676–89. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
