

Admixture mapping comes of age
- PMID: 20594047
- PMCID: PMC7454031
- DOI: 10.1146/annurev-genom-082509-141523
Admixture mapping comes of age
Abstract
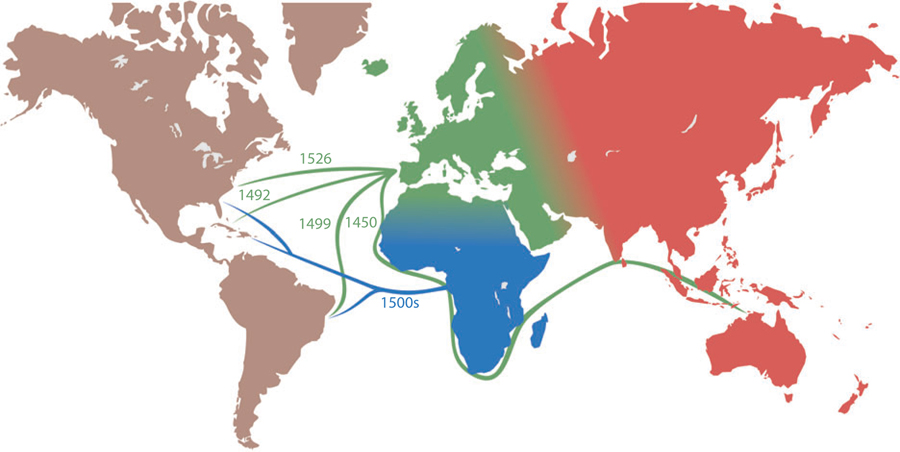
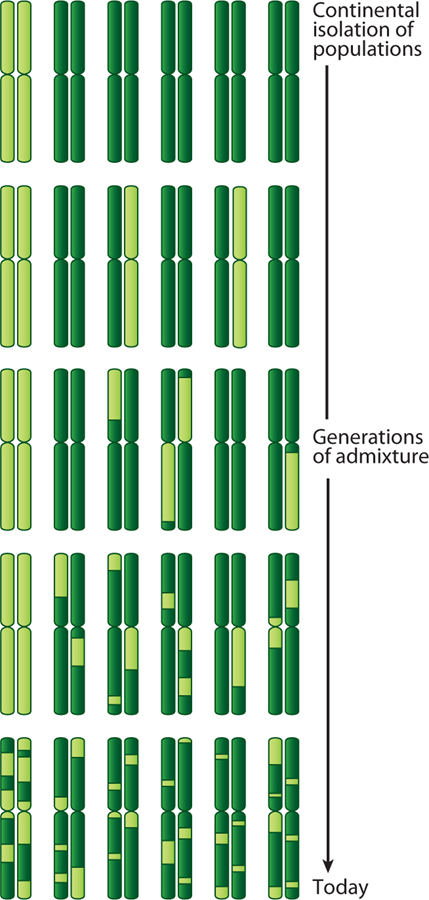
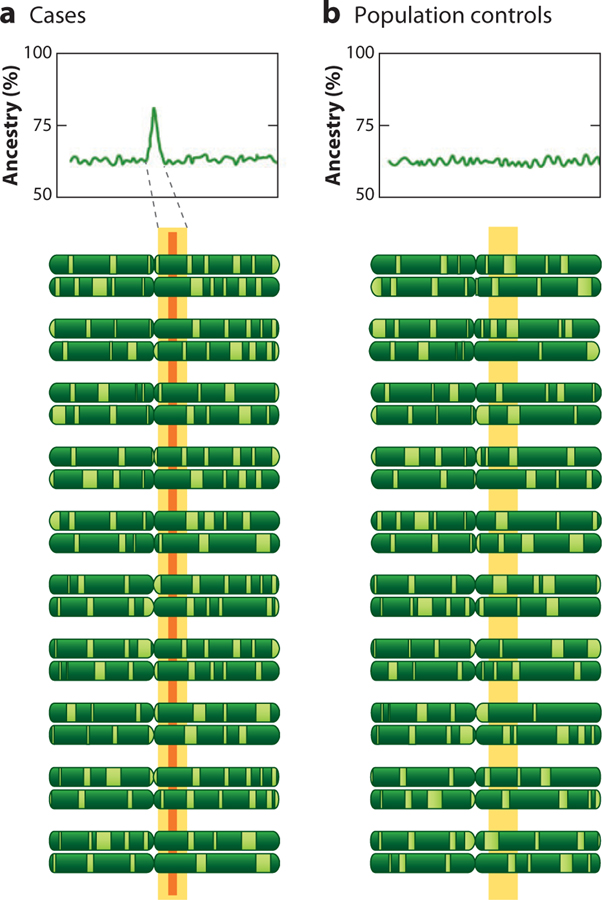
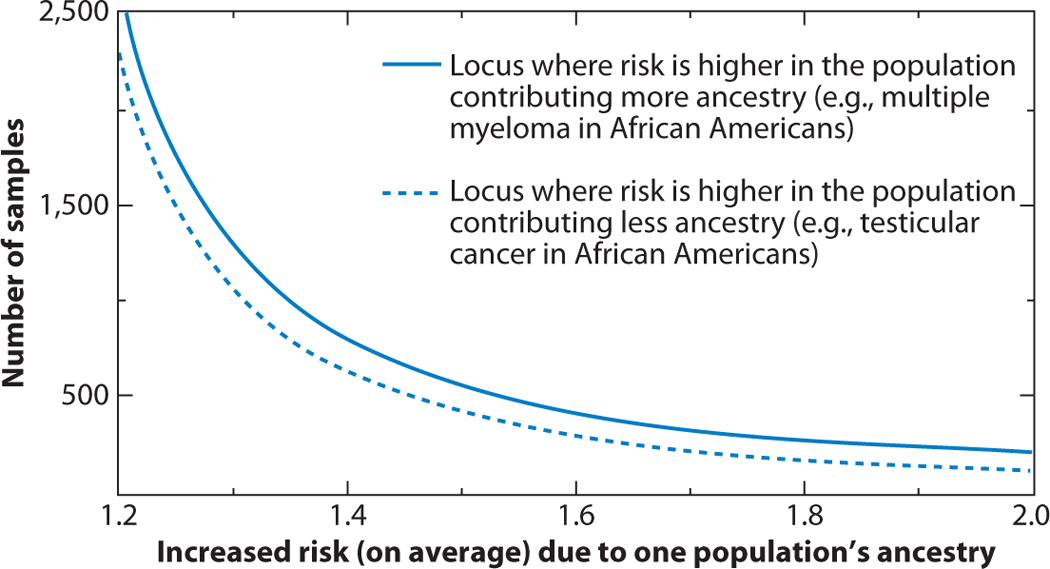
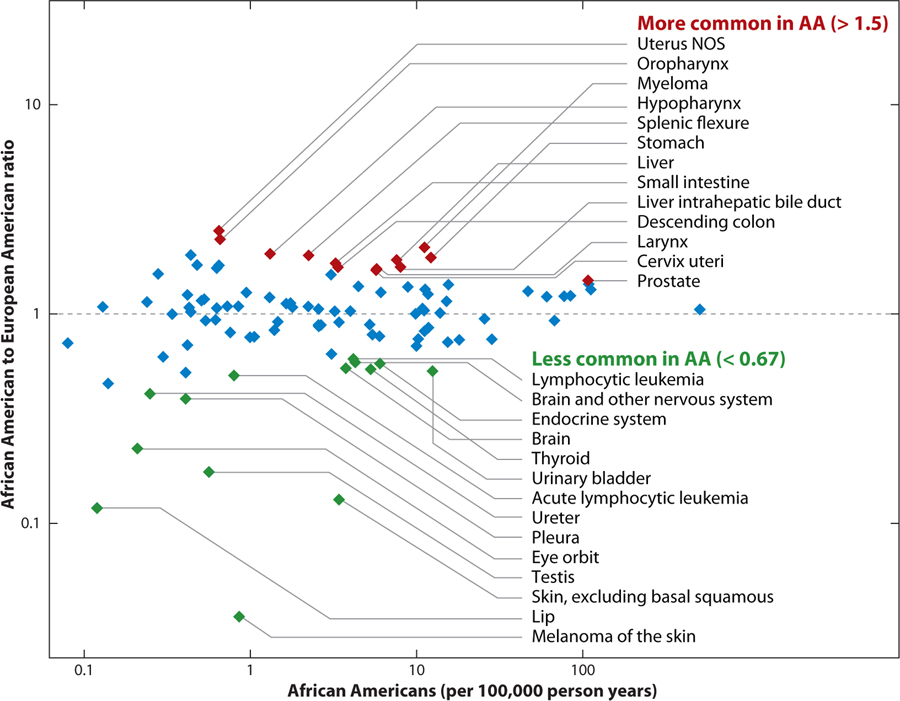
Admixture mapping is based on the hypothesis that differences in disease rates between populations are due in part to frequency differences in disease-causing genetic variants. In admixed populations, these genetic variants occur more often on chromosome segments inherited from the ancestral population with the higher disease variant frequency. A genome scan for disease association requires only enough markers to identify the ancestral chromosome segments; for recently admixed populations, such as African Americans, 1,500-2,500 ancestry-informative markers (AIMs) are sufficient. The method was proposed over 50 years ago, but the AIM panels and statistical methods required have only recently become available. Since the first admixture scan in 2005, the genetic bases for a range of diseases/traits have been identified by admixture mapping. Here, we provide a historical perspective, review AIM panels and software packages, and discuss recent successes and unexpected insights into human diseases that exhibit disparate rates across human populations.
Figures
References
-
- Althuis MD, Brogan DD, Coates RJ, Daling JR, Gammon MD, et al. 2003. Breast cancers among very young premenopausal women (United States). Cancer Causes Control 14:151–60 - PubMed
-
- Amundadottir LT, Sulem P, Gudmundsson J, Helgason A, Baker A, et al. 2006. A common variant associated with prostate cancer in European and African populations. Nat. Genet. 38:652–8 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
