

Knowledge-based annotation of small molecule binding sites in proteins
- PMID: 20594344
- PMCID: PMC2909224
- DOI: 10.1186/1471-2105-11-365
Knowledge-based annotation of small molecule binding sites in proteins
Abstract
Background: The study of protein-small molecule interactions is vital for understanding protein function and for practical applications in drug discovery. To benefit from the rapidly increasing structural data, it is essential to improve the tools that enable large scale binding site prediction with greater emphasis on their biological validity.
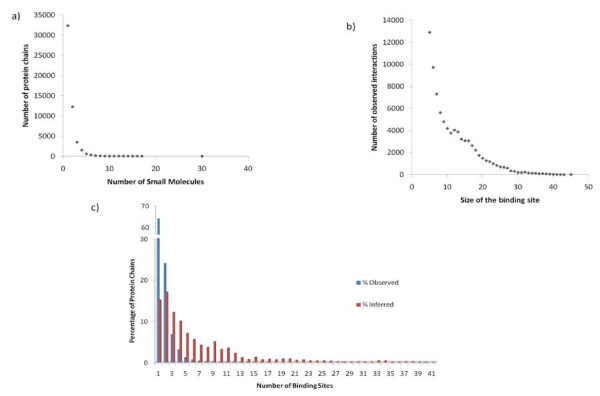
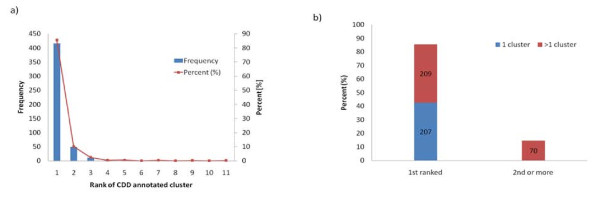
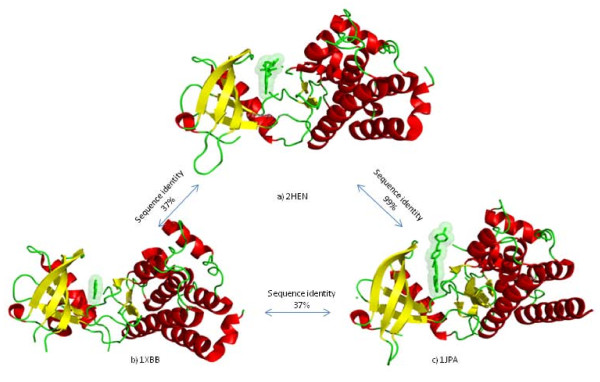
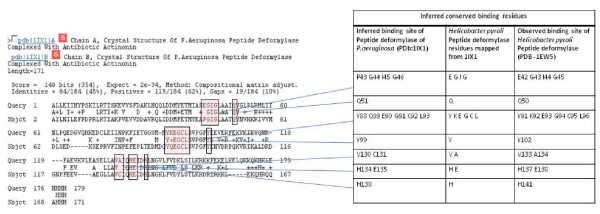
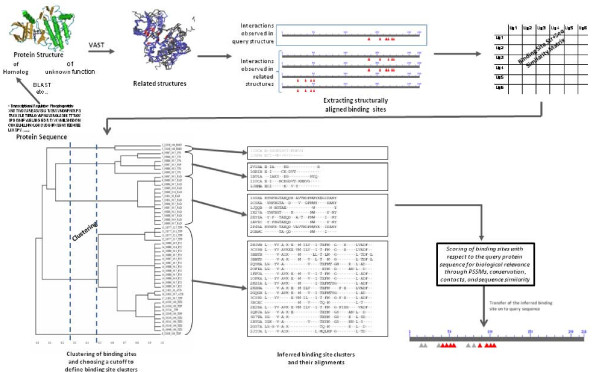
Results: We have developed a new method for the annotation of protein-small molecule binding sites, using inference by homology, which allows us to extend annotation onto protein sequences without experimental data available. To ensure biological relevance of binding sites, our method clusters similar binding sites found in homologous protein structures based on their sequence and structure conservation. Binding sites which appear evolutionarily conserved among non-redundant sets of homologous proteins are given higher priority. After binding sites are clustered, position specific score matrices (PSSMs) are constructed from the corresponding binding site alignments. Together with other measures, the PSSMs are subsequently used to rank binding sites to assess how well they match the query and to better gauge their biological relevance. The method also facilitates a succinct and informative representation of observed and inferred binding sites from homologs with known three-dimensional structures, thereby providing the means to analyze conservation and diversity of binding modes. Furthermore, the chemical properties of small molecules bound to the inferred binding sites can be used as a starting point in small molecule virtual screening. The method was validated by comparison to other binding site prediction methods and to a collection of manually curated binding site annotations. We show that our method achieves a sensitivity of 72% at predicting biologically relevant binding sites and can accurately discriminate those sites that bind biological small molecules from non-biological ones.
Conclusions: A new algorithm has been developed to predict binding sites with high accuracy in terms of their biological validity. It also provides a common platform for function prediction, knowledge-based docking and for small molecule virtual screening. The method can be applied even for a query sequence without structure. The method is available at http://www.ncbi.nlm.nih.gov/Structure/ibis/ibis.cgi.
Figures
Similar articles
-
Domain-based small molecule binding site annotation.BMC Bioinformatics. 2006 Mar 17;7:152. doi: 10.1186/1471-2105-7-152. BMC Bioinformatics. 2006. PMID: 16545112 Free PMC article.
-
Homology inference of protein-protein interactions via conserved binding sites.PLoS One. 2012;7(1):e28896. doi: 10.1371/journal.pone.0028896. Epub 2012 Jan 31. PLoS One. 2012. PMID: 22303436 Free PMC article.
-
Inferred Biomolecular Interaction Server--a web server to analyze and predict protein interacting partners and binding sites.Nucleic Acids Res. 2010 Jan;38(Database issue):D518-24. doi: 10.1093/nar/gkp842. Epub 2009 Oct 20. Nucleic Acids Res. 2010. PMID: 19843613 Free PMC article.
-
Computational approaches for protein function prediction: a combined strategy from multiple sequence alignment to molecular docking-based virtual screening.Biochim Biophys Acta. 2010 Sep;1804(9):1695-712. doi: 10.1016/j.bbapap.2010.04.008. Epub 2010 Apr 28. Biochim Biophys Acta. 2010. PMID: 20433957 Review.
-
Binding site matching in rational drug design: algorithms and applications.Brief Bioinform. 2019 Nov 27;20(6):2167-2184. doi: 10.1093/bib/bby078. Brief Bioinform. 2019. PMID: 30169563 Free PMC article. Review.
Cited by
-
Modulating protein-protein interactions with small molecules: the importance of binding hotspots.J Mol Biol. 2012 Jan 13;415(2):443-53. doi: 10.1016/j.jmb.2011.12.026. Epub 2011 Dec 16. J Mol Biol. 2012. PMID: 22198293 Free PMC article.
-
Systems pharmacology: network analysis to identify multiscale mechanisms of drug action.Annu Rev Pharmacol Toxicol. 2012;52:505-21. doi: 10.1146/annurev-pharmtox-010611-134520. Annu Rev Pharmacol Toxicol. 2012. PMID: 22235860 Free PMC article. Review.
-
Simplified sequence-based method for ATP-binding prediction using contextual local evolutionary conservation.Algorithms Mol Biol. 2014 Mar 11;9(1):7. doi: 10.1186/1748-7188-9-7. Algorithms Mol Biol. 2014. PMID: 24618258 Free PMC article.
-
Computational large-scale mapping of protein-protein interactions using structural complexes.Curr Protoc Protein Sci. 2013 Sep 24;73:3.9.1-3.9.9. doi: 10.1002/0471140864.ps0309s73. Curr Protoc Protein Sci. 2013. PMID: 24510594 Free PMC article.
-
HemeBIND: a novel method for heme binding residue prediction by combining structural and sequence information.BMC Bioinformatics. 2011 May 26;12:207. doi: 10.1186/1471-2105-12-207. BMC Bioinformatics. 2011. PMID: 21612668 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
