

Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia--MLASA syndrome
- PMID: 20598274
- PMCID: PMC2896778
- DOI: 10.1016/j.ajhg.2010.06.001
Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia--MLASA syndrome
Abstract
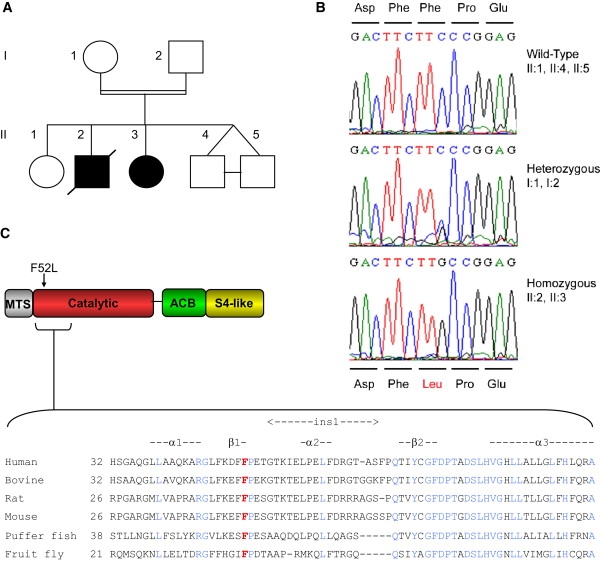
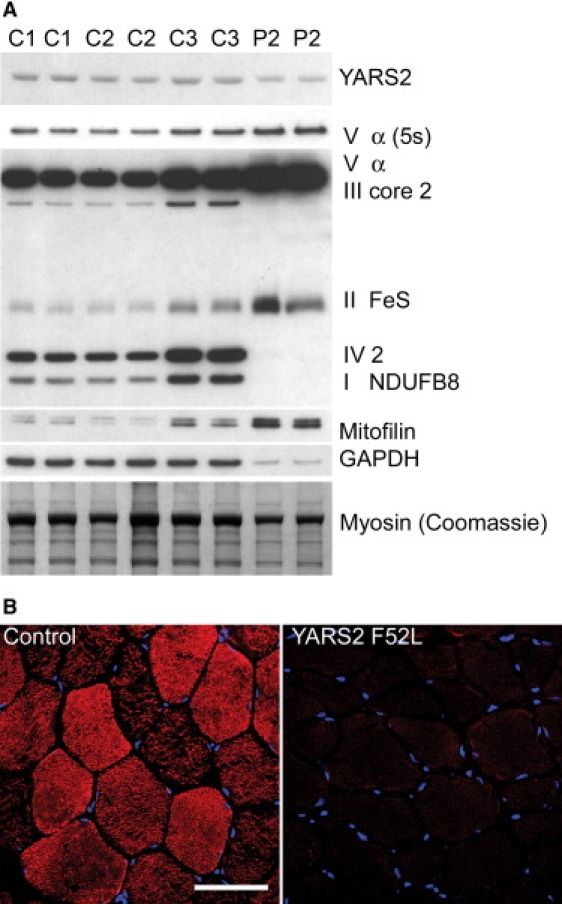
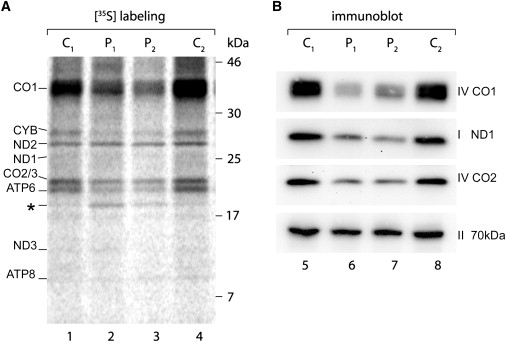
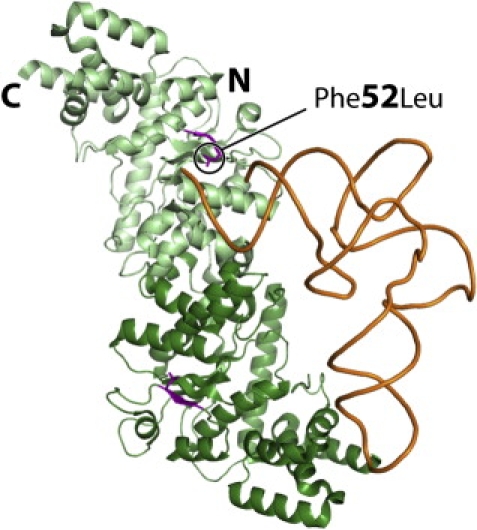
Mitochondrial respiratory chain disorders are a heterogeneous group of disorders in which the underlying genetic defect is often unknown. We have identified a pathogenic mutation (c.156C>G [p.F52L]) in YARS2, located at chromosome 12p11.21, by using genome-wide SNP-based homozygosity analysis of a family with affected members displaying myopathy, lactic acidosis, and sideroblastic anemia (MLASA). We subsequently identified the same mutation in another unrelated MLASA patient. The YARS2 gene product, mitochondrial tyrosyl-tRNA synthetase (YARS2), was present at lower levels in skeletal muscle whereas fibroblasts were relatively normal. Complex I, III, and IV were dysfunctional as indicated by enzyme analysis, immunoblotting, and immunohistochemistry. A mitochondrial protein-synthesis assay showed reduced levels of respiratory chain subunits in myotubes generated from patient cell lines. A tRNA aminoacylation assay revealed that mutant YARS2 was still active; however, enzyme kinetics were abnormal compared to the wild-type protein. We propose that the reduced aminoacylation activity of mutant YARS2 enzyme leads to decreased mitochondrial protein synthesis, resulting in mitochondrial respiratory chain dysfunction. MLASA has previously been associated with PUS1 mutations; hence, the YARS2 mutation reported here is an alternative cause of MLASA.
Copyright 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
A distinct mitochondrial myopathy, lactic acidosis and sideroblastic anemia (MLASA) phenotype associates with YARS2 mutations.Am J Med Genet A. 2013 Sep;161A(9):2334-8. doi: 10.1002/ajmg.a.36065. Epub 2013 Aug 5. Am J Med Genet A. 2013. PMID: 23918765 Free PMC article.
-
A novel mutation in YARS2 causes myopathy with lactic acidosis and sideroblastic anemia.Hum Mutat. 2012 Aug;33(8):1201-6. doi: 10.1002/humu.22098. Epub 2012 May 7. Hum Mutat. 2012. PMID: 22504945
-
Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia.Orphanet J Rare Dis. 2013 Dec 17;8:193. doi: 10.1186/1750-1172-8-193. Orphanet J Rare Dis. 2013. PMID: 24344687 Free PMC article.
-
A Novel PUS1 Mutation in 2 Siblings with MLASA Syndrome: A Review of the Literature.J Pediatr Hematol Oncol. 2021 May 1;43(4):e592-e595. doi: 10.1097/MPH.0000000000001806. J Pediatr Hematol Oncol. 2021. PMID: 32287105 Review.
-
Pathophysiology and genetic mutations in congenital sideroblastic anemia.Pediatr Int. 2013 Dec;55(6):675-9. doi: 10.1111/ped.12217. Pediatr Int. 2013. PMID: 24003969 Review.
Cited by
-
Haematological abnormalities in mitochondrial disorders.Singapore Med J. 2015 Jul;56(7):412-9. doi: 10.11622/smedj.2015112. Singapore Med J. 2015. PMID: 26243978 Free PMC article.
-
RMND1 deficiency associated with neonatal lactic acidosis, infantile onset renal failure, deafness, and multiorgan involvement.Eur J Hum Genet. 2015 Oct;23(10):1301-7. doi: 10.1038/ejhg.2014.293. Epub 2015 Jan 21. Eur J Hum Genet. 2015. PMID: 25604853 Free PMC article.
-
Mutation analysis of Chinese sporadic congenital sideroblastic anemia by targeted capture sequencing.J Hematol Oncol. 2015 May 20;8:55. doi: 10.1186/s13045-015-0154-0. J Hematol Oncol. 2015. PMID: 25985931 Free PMC article.
-
YARS2 Missense Variant in Belgian Shepherd Dogs with Cardiomyopathy and Juvenile Mortality.Genes (Basel). 2020 Mar 14;11(3):313. doi: 10.3390/genes11030313. Genes (Basel). 2020. PMID: 32183361 Free PMC article.
-
The crystal structure of human GlnRS provides basis for the development of neurological disorders.Nucleic Acids Res. 2016 Apr 20;44(7):3420-31. doi: 10.1093/nar/gkw082. Epub 2016 Feb 10. Nucleic Acids Res. 2016. PMID: 26869582 Free PMC article.
References
-
- Skladal D., Halliday J., Thorburn D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. - PubMed
-
- Thorburn D. Practical problems in detecting abnormal mitochondrial function and genomes. Hum. Reprod. 2000;15 (Suppl):57–67. - PubMed
-
- DiMauro S., Schon E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003;348:2656–2668. - PubMed
-
- Zeharia A., Fischel-Ghodsian N., Casas K., Bykhocskaya Y., Tamari H., Lev D., Mimouni M., Lerman-Sagie T. Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: An autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. J. Child Neurol. 2005;20:449–452. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
