

A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex
- PMID: 20603002
- PMCID: PMC3655264
- DOI: 10.1016/j.cell.2010.05.006
A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex
Abstract
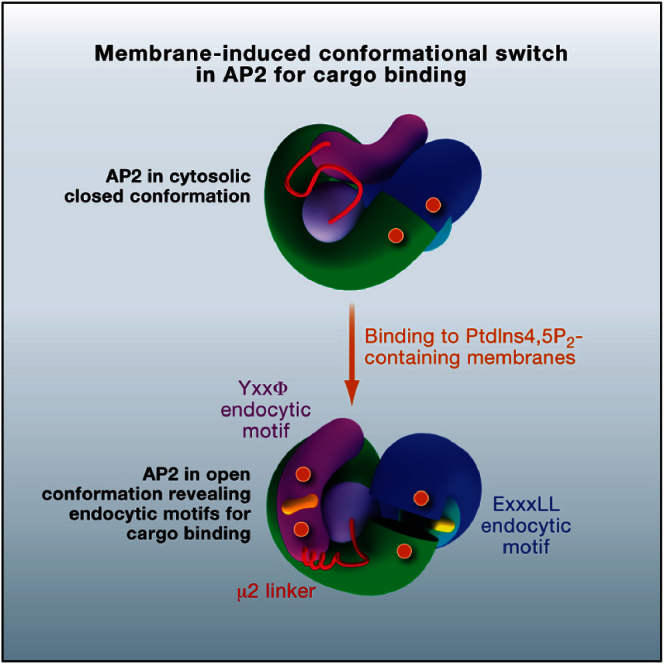
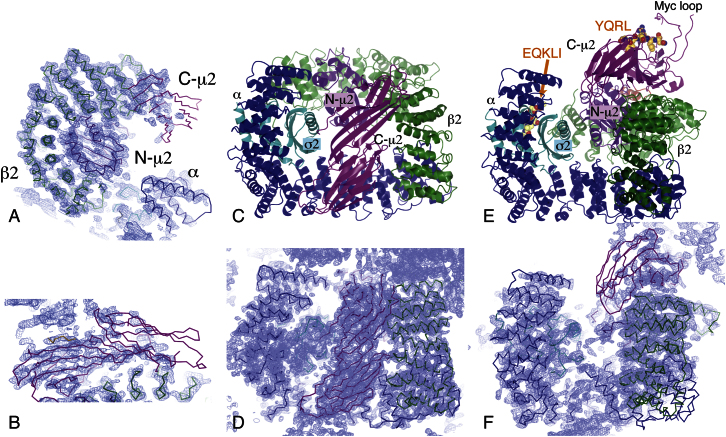
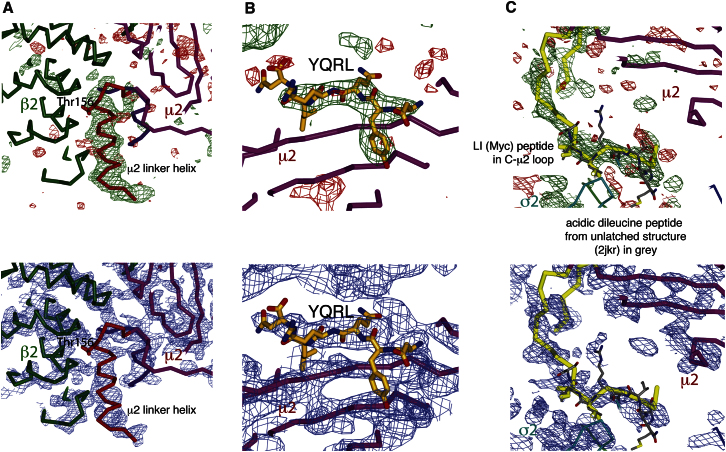
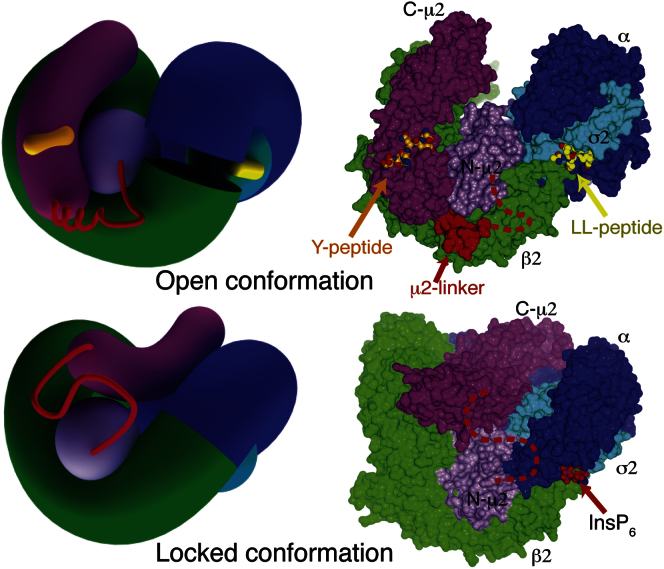
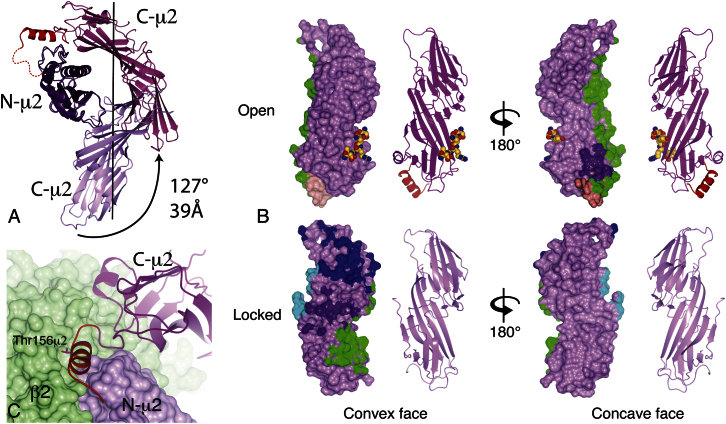
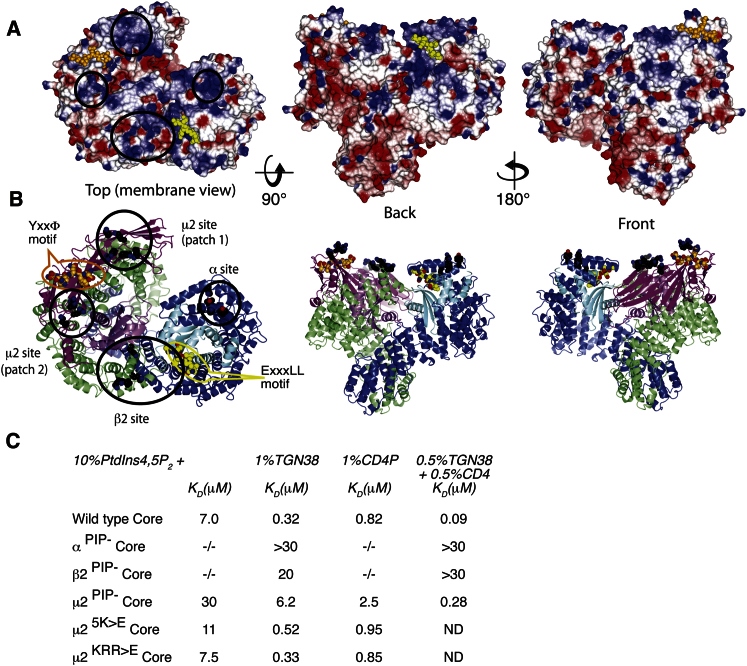
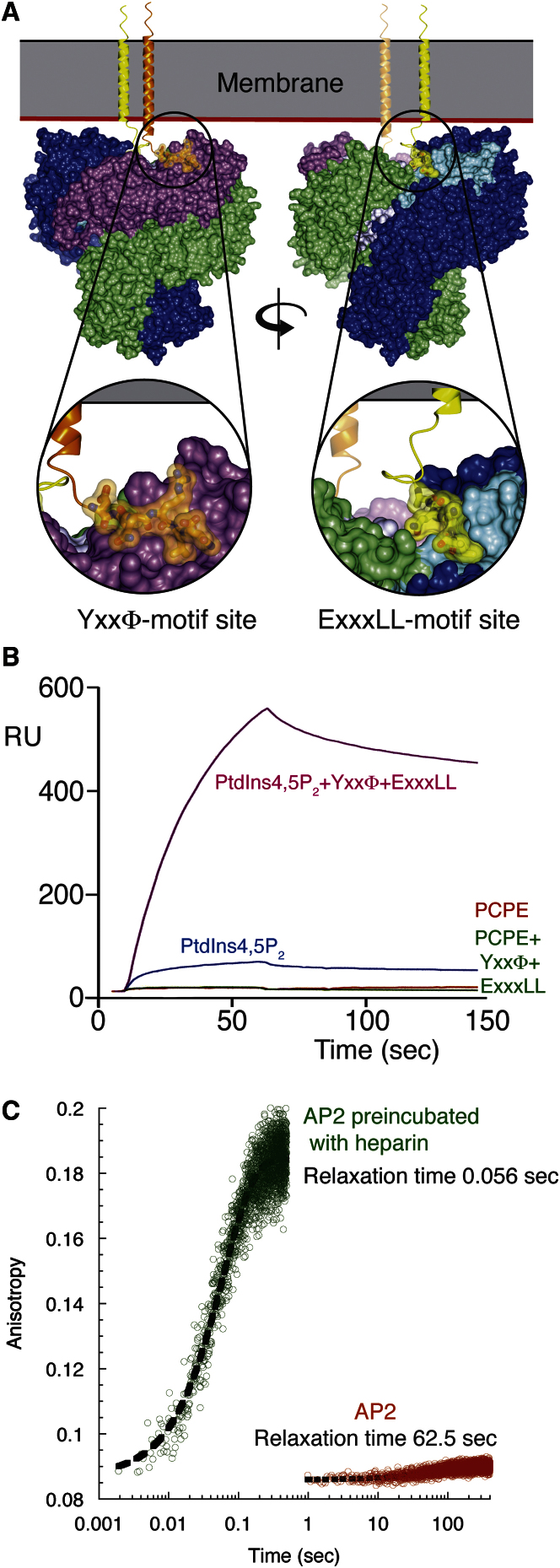
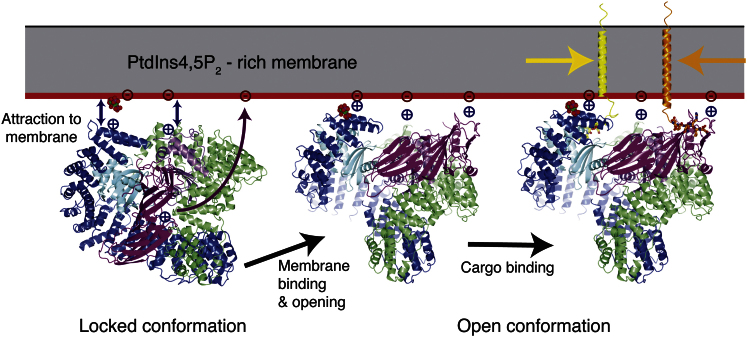
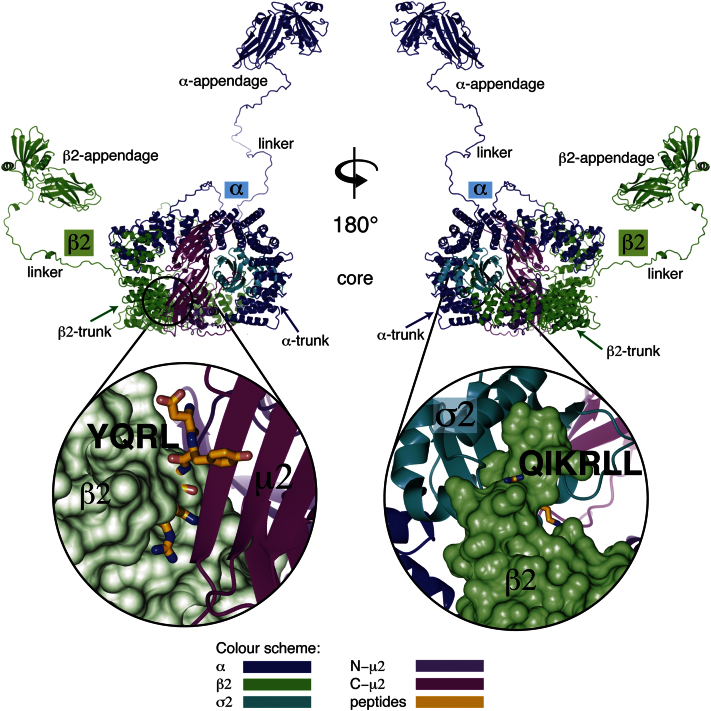
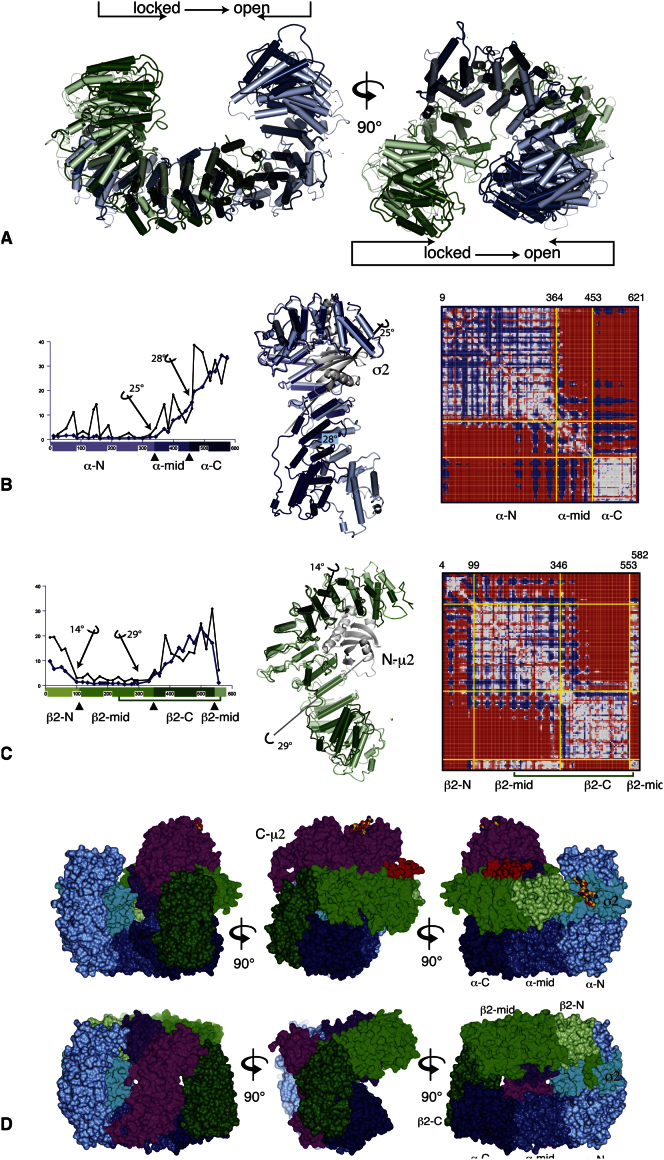
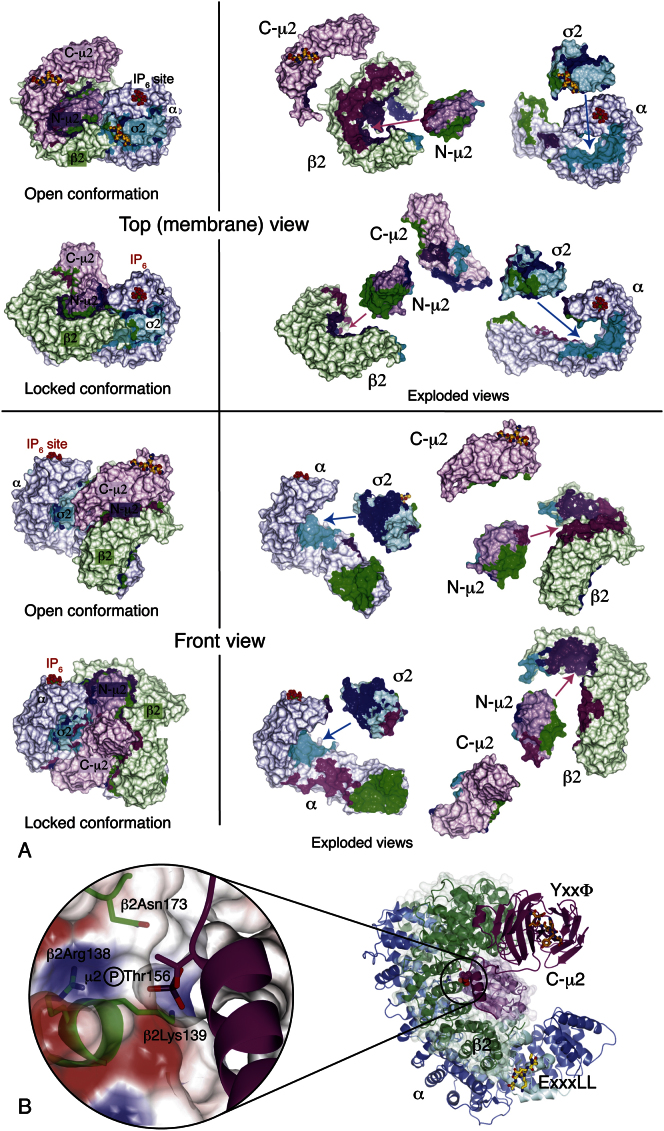
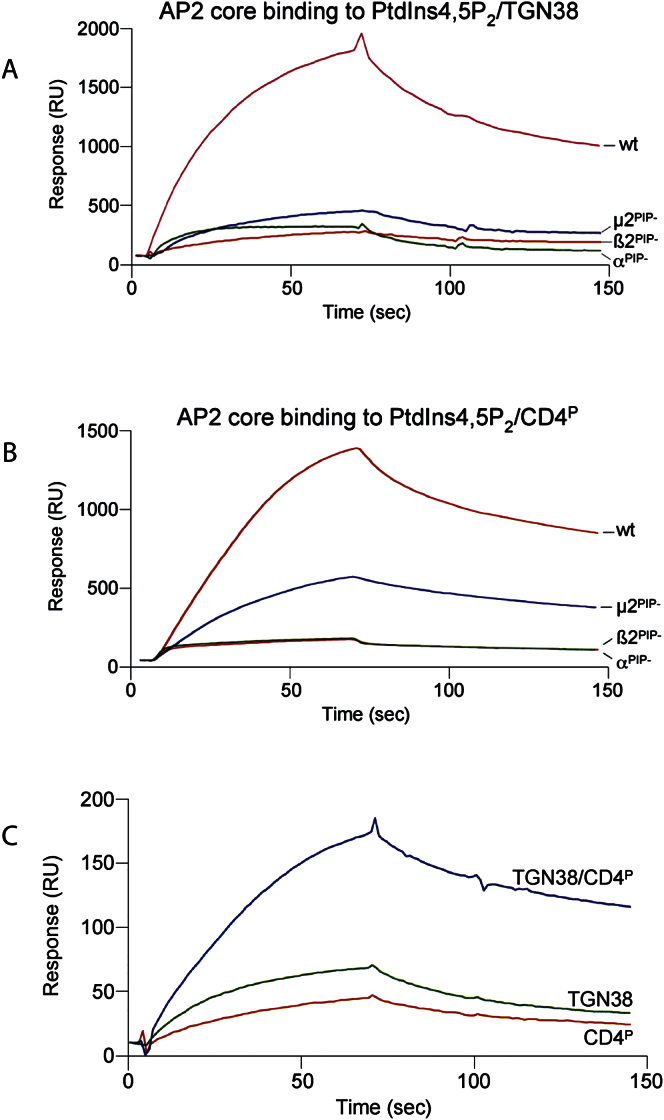
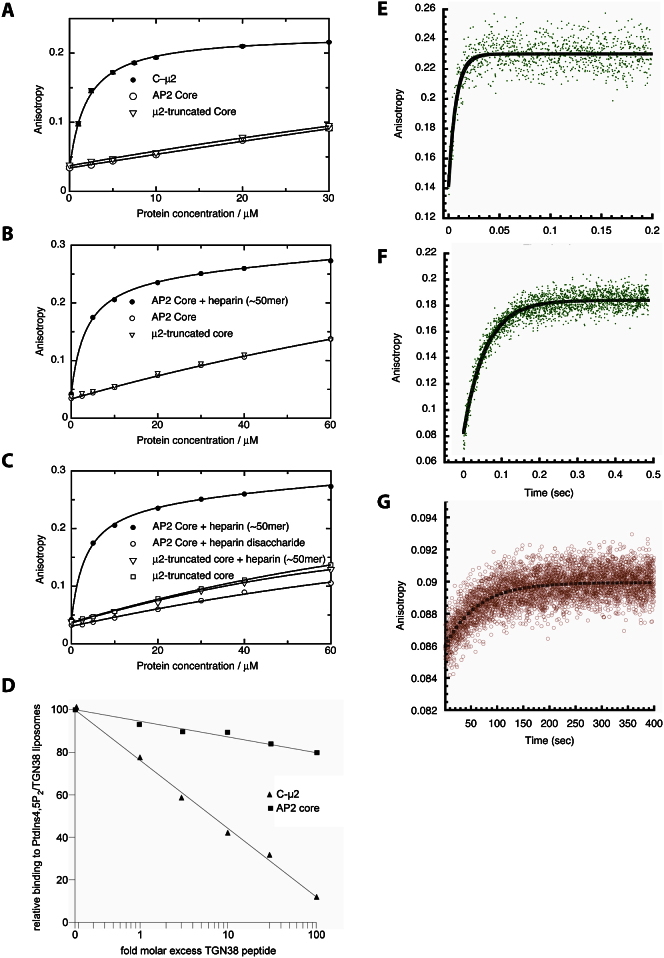
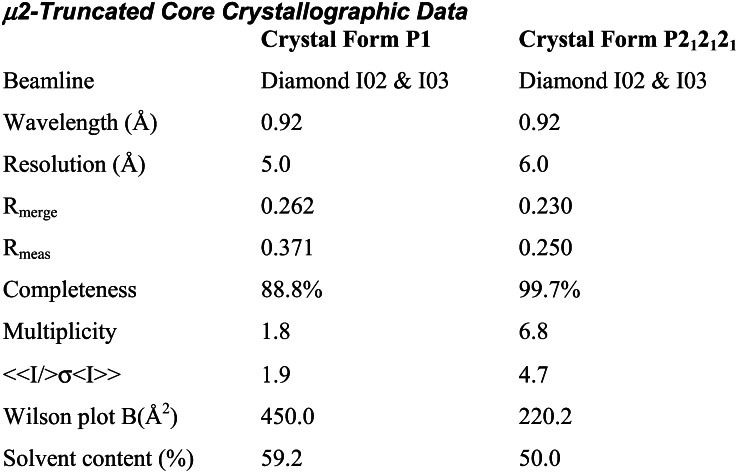
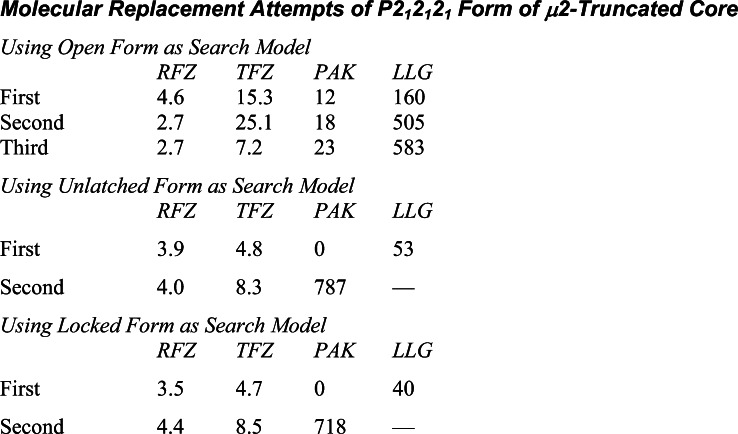
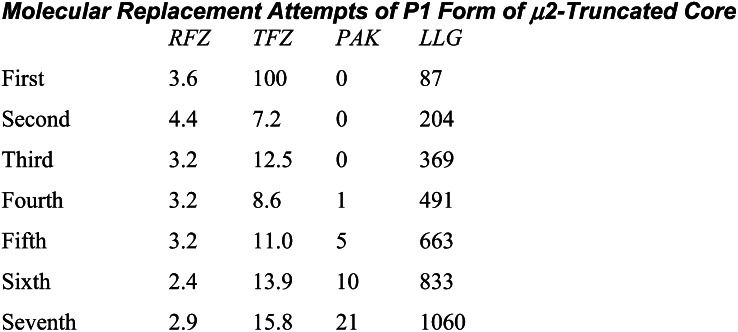
The AP2 adaptor complex (alpha, beta2, sigma2, and mu2 subunits) crosslinks the endocytic clathrin scaffold to PtdIns4,5P(2)-containing membranes and transmembrane protein cargo. In the "locked" cytosolic form, AP2's binding sites for the two endocytic motifs, YxxPhi on the C-terminal domain of mu2 (C-mu2) and [ED]xxxL[LI] on sigma2, are blocked by parts of beta2. Using protein crystallography, we show that AP2 undergoes a large conformational change in which C-mu2 relocates to an orthogonal face of the complex, simultaneously unblocking both cargo-binding sites; the previously unstructured mu2 linker becomes helical and binds back onto the complex. This structural rearrangement results in AP2's four PtdIns4,5P(2)- and two endocytic motif-binding sites becoming coplanar, facilitating their simultaneous interaction with PtdIns4,5P(2)/cargo-containing membranes. Using a range of biophysical techniques, we show that the endocytic cargo binding of AP2 is driven by its interaction with PtdIns4,5P(2)-containing membranes.
Figures
References
-
- Austin C., Hinners I., Tooze S.A. Direct and GTP-dependent interaction of ADP-ribosylation factor 1 with clathrin adaptor protein AP-1 on immature secretory granules. J. Biol. Chem. 2000;275:21862–21869. - PubMed
-
- Bonifacino J.S., Traub L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003;72:395–447. - PubMed
Supplemental References
-
- Abrahams, J.P. (1997). Bias reduction in phase refinement by modified interference functions: introducing the γ function. Acta Crystallogr. D Biol. Crystallogr. 53, 371–376. - PubMed
-
- Adams, P.D., Grosse-Kunstleve, R.W., Hung, L.W., Ioerger, T.R., McCoy, A.J., Moriarty, N.W., Read, R.J., Sacchettini, J.C., Sauter, N.K., and Terwilliger, T.C. (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954. - PubMed
-
- Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M., and Paciorek, W. (2003). Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 59, 2023–2030. - PubMed
-
- Collins, B.M., McCoy, A.J., Kent, H.M., Evans, P.R., and Owen, D.J. (2002). Molecular architecture and functional model of the endocytic AP2 complex. Cell 109, 523–535. - PubMed
-
- Emsley, P., and Cowtan, K. (2004). Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
