

A comprehensive comparison of transmembrane domains reveals organelle-specific properties
- PMID: 20603021
- PMCID: PMC2928124
- DOI: 10.1016/j.cell.2010.05.037
A comprehensive comparison of transmembrane domains reveals organelle-specific properties
Abstract
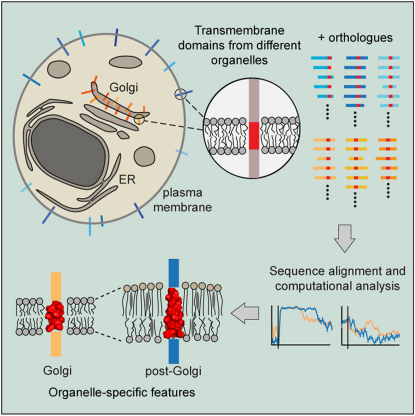
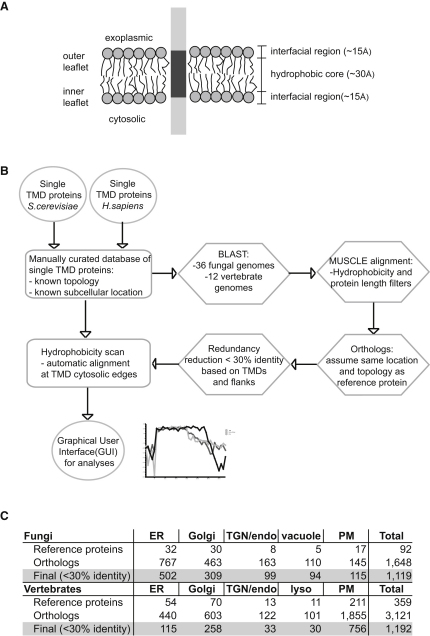
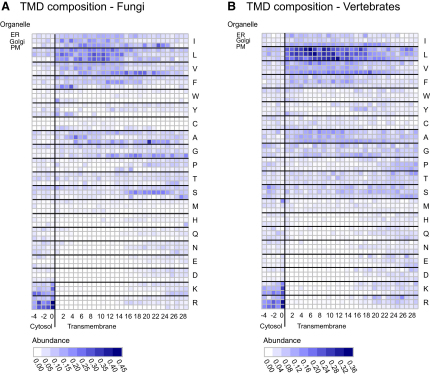
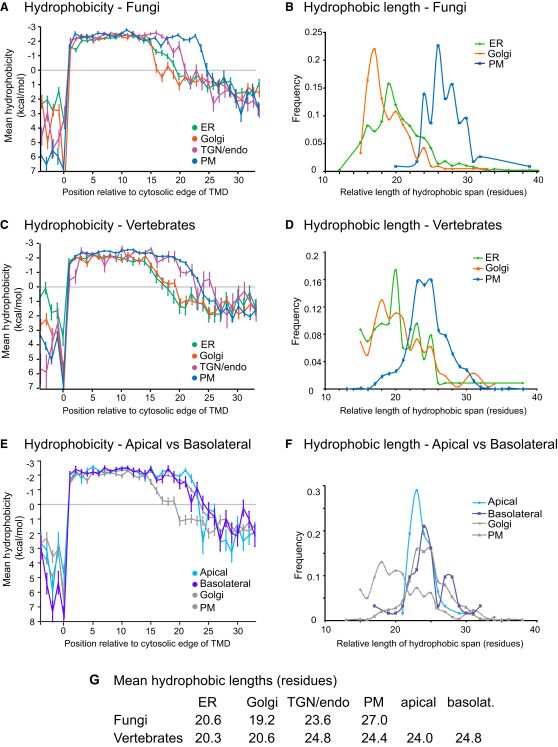
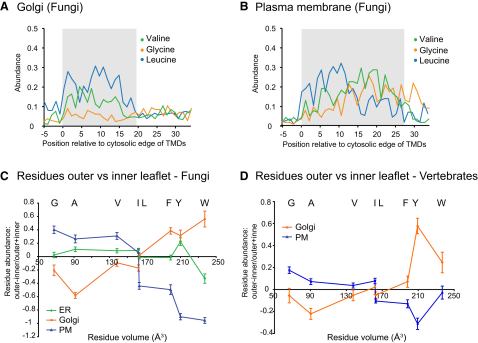
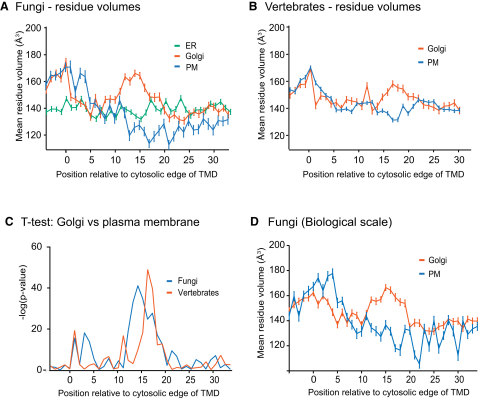
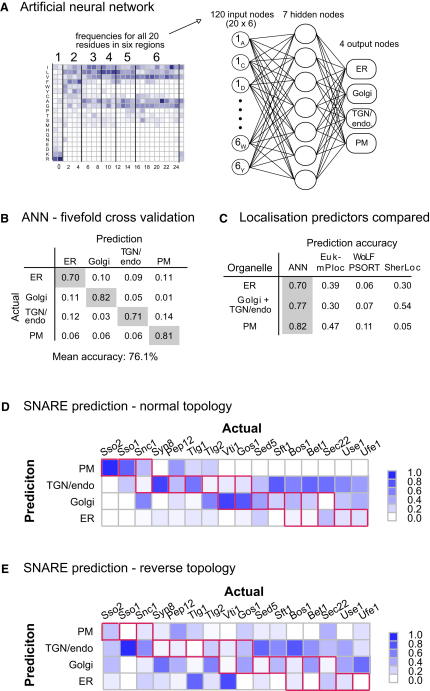
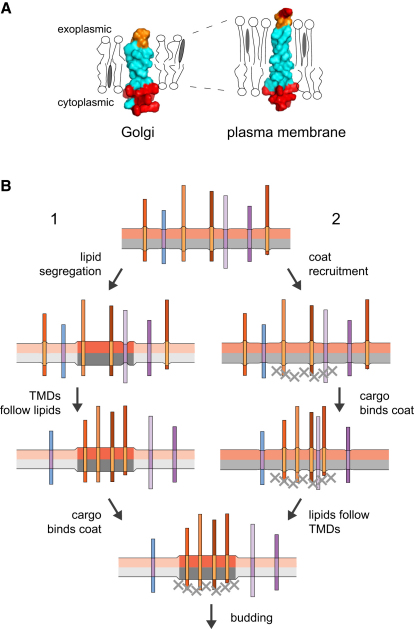
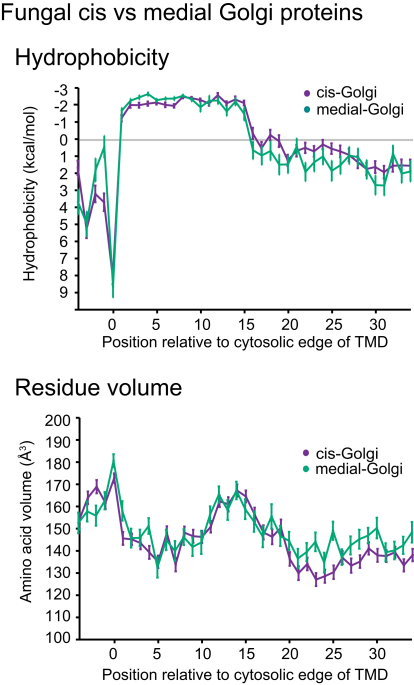
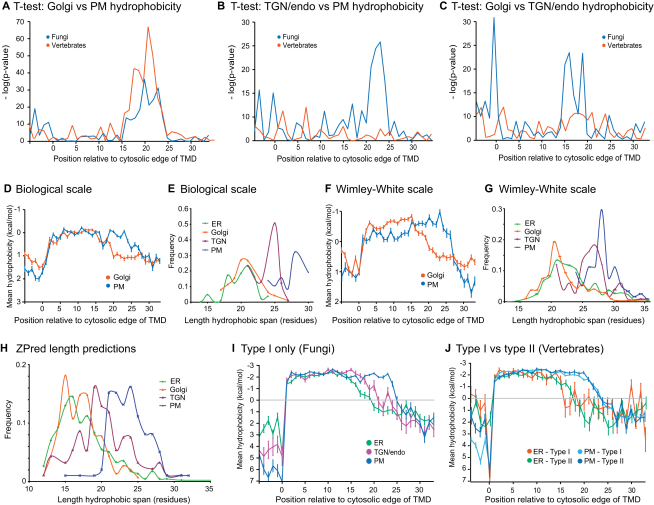
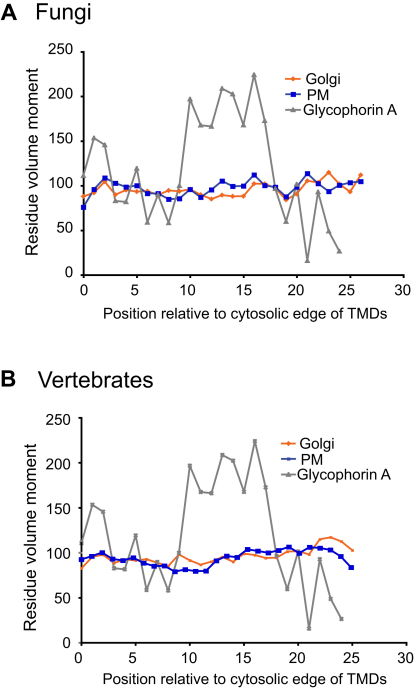
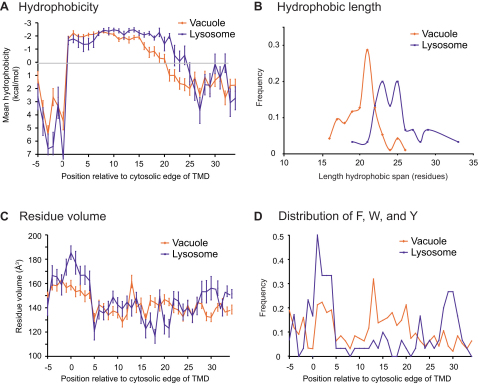
The various membranes of eukaryotic cells differ in composition, but it is at present unclear if this results in differences in physical properties. The sequences of transmembrane domains (TMDs) of integral membrane proteins should reflect the physical properties of the bilayers in which they reside. We used large datasets from both fungi and vertebrates to perform a comprehensive comparison of the TMDs of proteins from different organelles. We find that TMDs are not generic but have organelle-specific properties with a dichotomy in TMD length between the early and late parts of the secretory pathway. In addition, TMDs from post-ER organelles show striking asymmetries in amino acid compositions across the bilayer that is linked to residue size and varies between organelles. The pervasive presence of organelle-specific features among the TMDs of a particular organelle has implications for TMD prediction, regulation of protein activity by location, and sorting of proteins and lipids in the secretory pathway.
Copyright 2010 Elsevier Inc. All rights reserved.
Figures
References
-
- Andersen O.S., Koeppe R.E. Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007;36:107–130. - PubMed
-
- Bacia K., Schuette C.G., Kahya N., Jahn R., Schwille P. SNAREs prefer liquid-disordered over “raft” (liquid-ordered) domains when reconstituted into giant unilamellar vesicles. J. Biol. Chem. 2004;279:37951–37955. - PubMed
Supplemental References
-
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. - PubMed
-
- Chou, K.-C., and Shen, H.-B. (2007). Euk-mPLoc: a fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J. Proteome Res. 6, 1728–1734. - PubMed
-
- Decaens, C., Durand, M., Grosse, B., and Cassio, D. (2008). Which in vitro models could be best used to study hepatocyte polarity? Biol. Cell 100, 387–398. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
