

Can self-inhibitory peptides be derived from the interfaces of globular protein-protein interactions?
- PMID: 20607702
- PMCID: PMC2952690
- DOI: 10.1002/prot.22785
Can self-inhibitory peptides be derived from the interfaces of globular protein-protein interactions?
Abstract
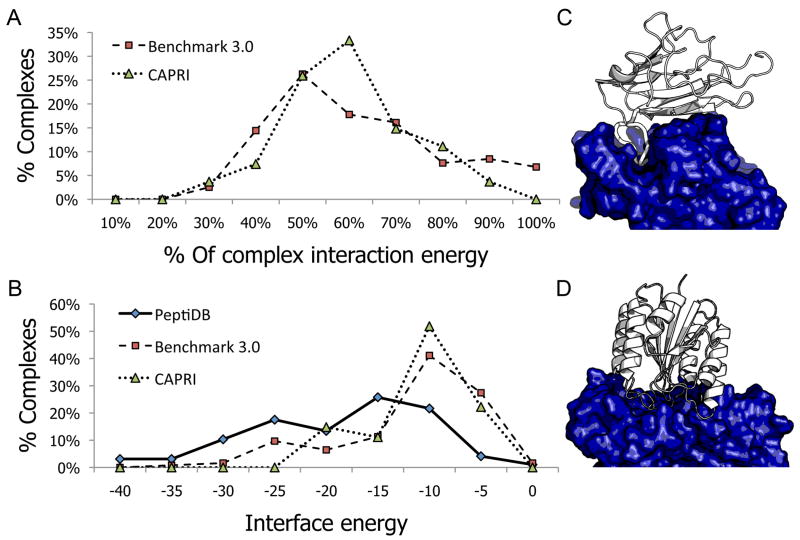
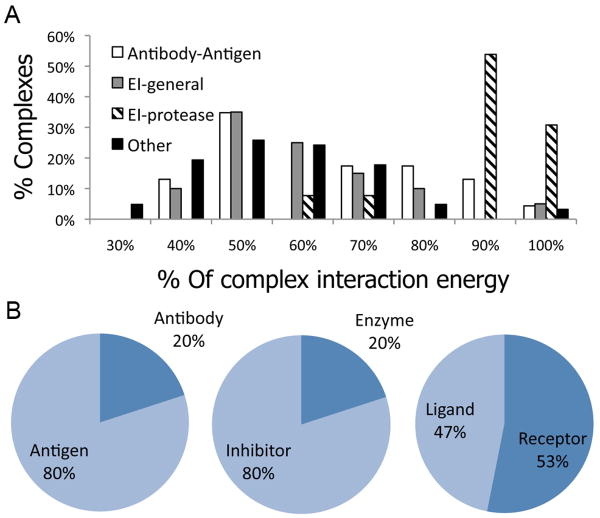
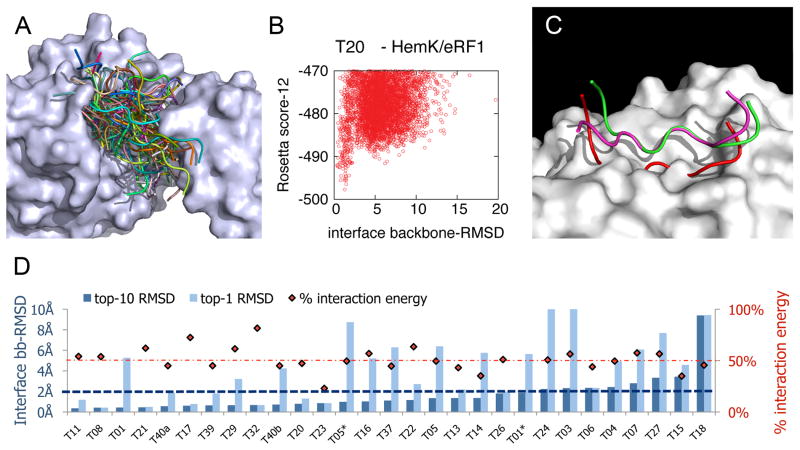
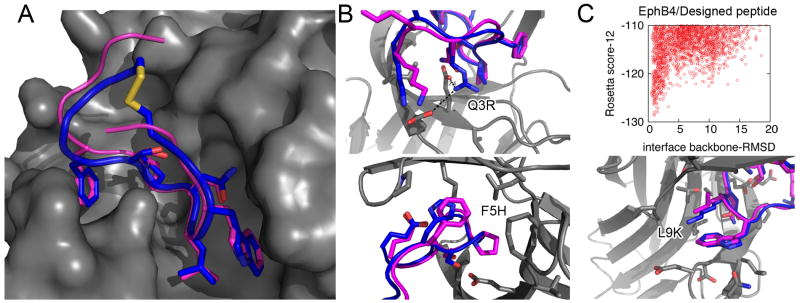
In this study, we assess on a large scale the possibility of deriving self-inhibitory peptides from protein domains with globular architectures. Such inhibitory peptides would inhibit interactions of their origin domain by mimicking its mode of binding to cognate partners, and could serve as promising leads for rational design of inhibitory drugs. For our large-scale analysis, we analyzed short linear segments that were cut out of protein interfaces in silico in complex structures of protein-protein docking Benchmark 3.0 and CAPRI targets from rounds 1-19. Our results suggest that more than 50% of these globular interactions are dominated by one short linear segment at the domain interface, which provides more than half of the original interaction energy. Importantly, in many cases the derived peptides show strong energetic preference for their original binding mode independently of the context of their original domain, as we demonstrate by extensive computational peptide docking experiments. As an in depth case study, we computationally design a candidate peptide to inhibit the EphB4-EphrinB2 interaction based on a short peptide derived from the G-H loop in EphrinB2. Altogether, we provide an elaborate framework for the in silico selection of candidate inhibitory molecules for protein-protein interactions. Such candidate molecules can be readily subjected to wet-laboratory experiments and provide highly promising starting points for subsequent drug design.
© 2010 Wiley-Liss, Inc.
Figures
References
-
- Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300(5618):445–452. - PubMed
-
- Petsalaki E, Russell RB. Peptide-mediated interactions in biological systems: new discoveries and applications. Curr Opin Biotechnol. 2008;19(4):344–350. - PubMed
-
- London N, Movshovitz-Attias D, Schueler-Furman O. The structural basis of peptide-protein binding strategies. Structure. 2010;18(2):188–199. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
