

Molecular genetics of bladder cancer: Emerging mechanisms of tumor initiation and progression
- PMID: 20610280
- PMCID: PMC2901550
- DOI: 10.1016/j.urolonc.2010.04.008
Molecular genetics of bladder cancer: Emerging mechanisms of tumor initiation and progression
Abstract
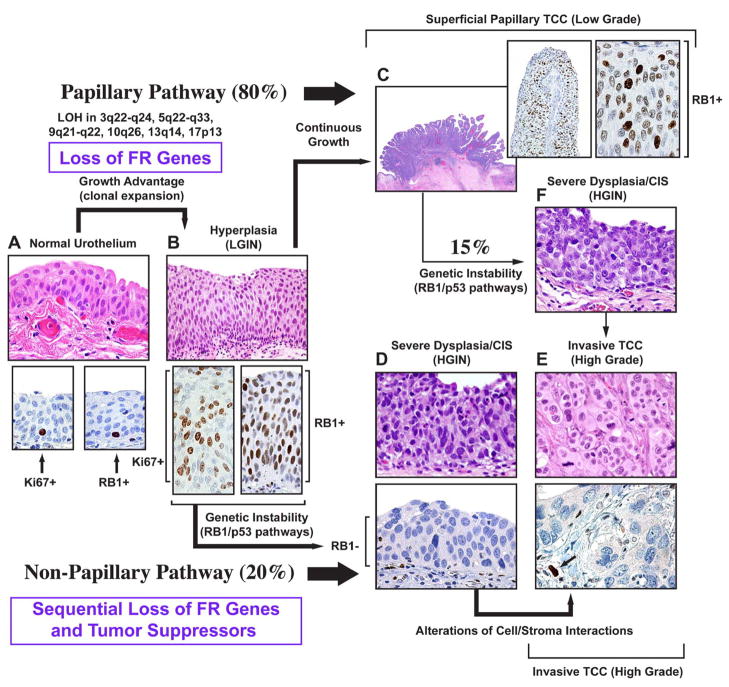
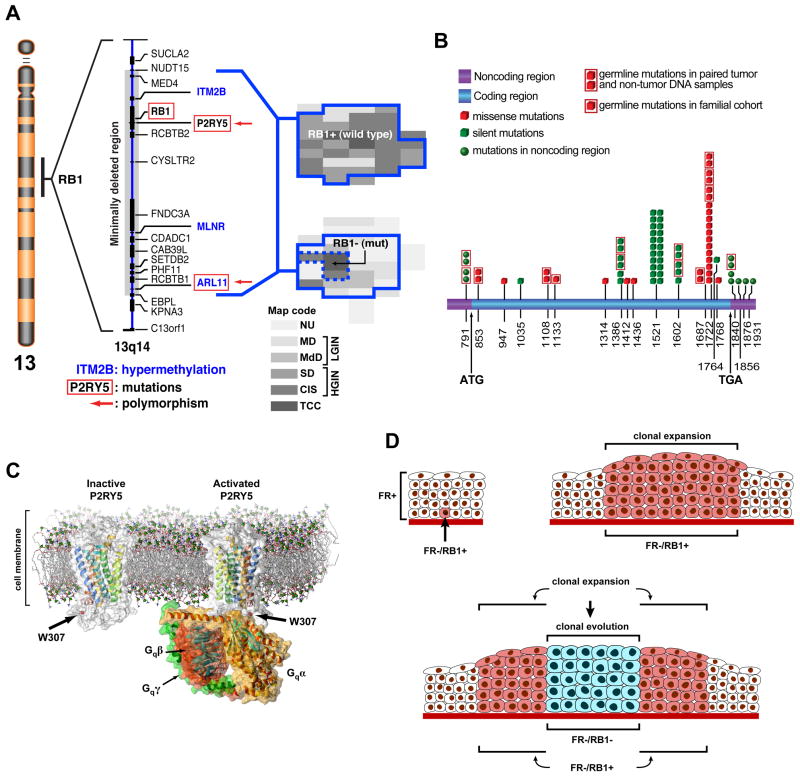
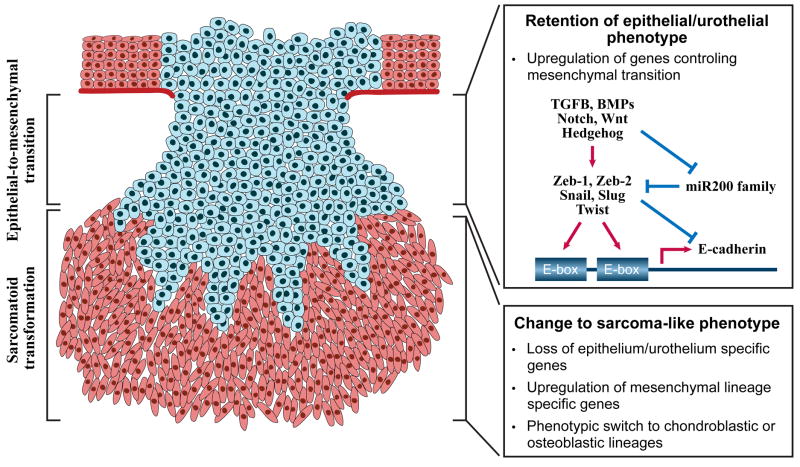
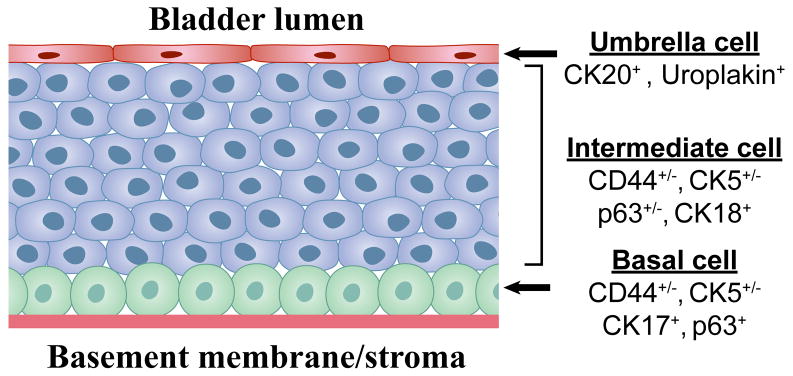
Urothelial cancer has served as one of the most important sources of information about the mutational events that underlie the development of human solid malignancies. Although "field effects" that affect the entire bladder mucosa appear to initiate disease, tumors develop along 2 distinct biological "tracks" that present vastly different challenges for clinical management. Recent whole genome methodologies have facilitated even more rapid progress in the identification of the molecular mechanisms involved in bladder cancer initiation and progression. Specifically, whole organ mapping combined with high resolution, high throughput SNP analyses have identified a novel class of candidate tumor suppressors ("forerunner genes") that localize near more familiar tumor suppressors but are disrupted at an earlier stage of cancer development. Furthermore, whole genome comparative genomic hybridization (CGH) and mRNA expression profiling have demonstrated that the 2 major subtypes of urothelial cancer (papillary/superficial and non-papillary/muscle-invasive) are truly distinct molecular entities, and in recent work our group has discovered that muscle-invasive tumors express molecular markers characteristic of a developmental process known as "epithelial-to-mesenchymal transition" (EMT). Emerging evidence indicates that urothelial cancers contain subpopulations of tumor-initiating cells ("cancer stem cells") but the phenotypes of these cells in different tumors are heterogeneous, raising questions about whether or not the 2 major subtypes of cancer share a common precursor. This review will provide an overview of these new insights and discuss priorities for future investigation.
Copyright 2010 Elsevier Inc. All rights reserved.
Figures
References
-
- Dinney CP, McConkey DJ, Millikan RE, et al. Focus on bladder cancer. Cancer Cell. 2004;6:111–6. - PubMed
-
- Olumi AF, Tsai YC, Nichols PW, et al. Allelic loss of chromosome 17p distinguishes high grade from low grade transitional cell carcinomas of the bladder. Cancer Res. 1990;50:7081–3. - PubMed
-
- Tsai YC, Nichols PW, Hiti AL, Williams Z, Skinner DG, Jones PA. Allelic losses of chromosomes 9, 11, and 17 in human bladder cancer. Cancer Res. 1990;50:44–7. - PubMed
-
- Ruppert JM, Tokino K, Sidransky D. Evidence for two bladder cancer suppressor loci on human chromosome 9. Cancer Res. 1993;53:5093–5. - PubMed
-
- Miyao N, Tsai YC, Lerner SP, et al. Role of chromosome 9 in human bladder cancer. Cancer Res. 1993;53:4066–70. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
