

Remarkable lethal G-to-A mutations in vif-proficient HIV-1 provirus by individual APOBEC3 proteins in humanized mice
- PMID: 20610708
- PMCID: PMC2937654
- DOI: 10.1128/JVI.00823-10
Remarkable lethal G-to-A mutations in vif-proficient HIV-1 provirus by individual APOBEC3 proteins in humanized mice
Abstract
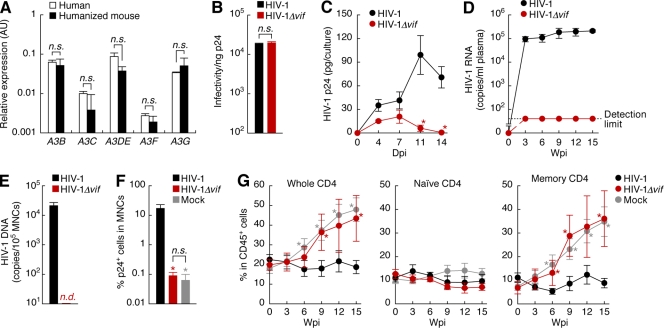
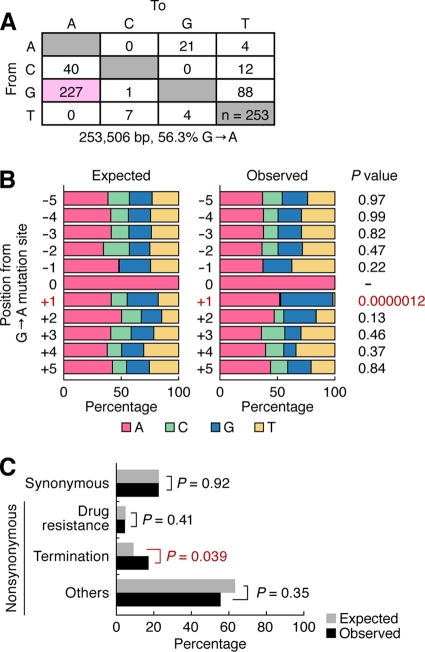
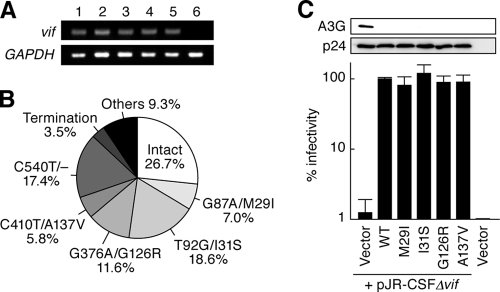
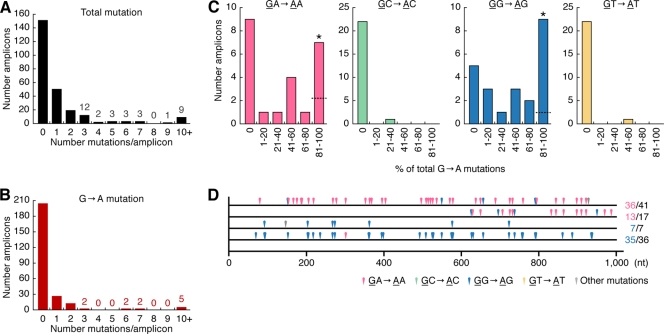
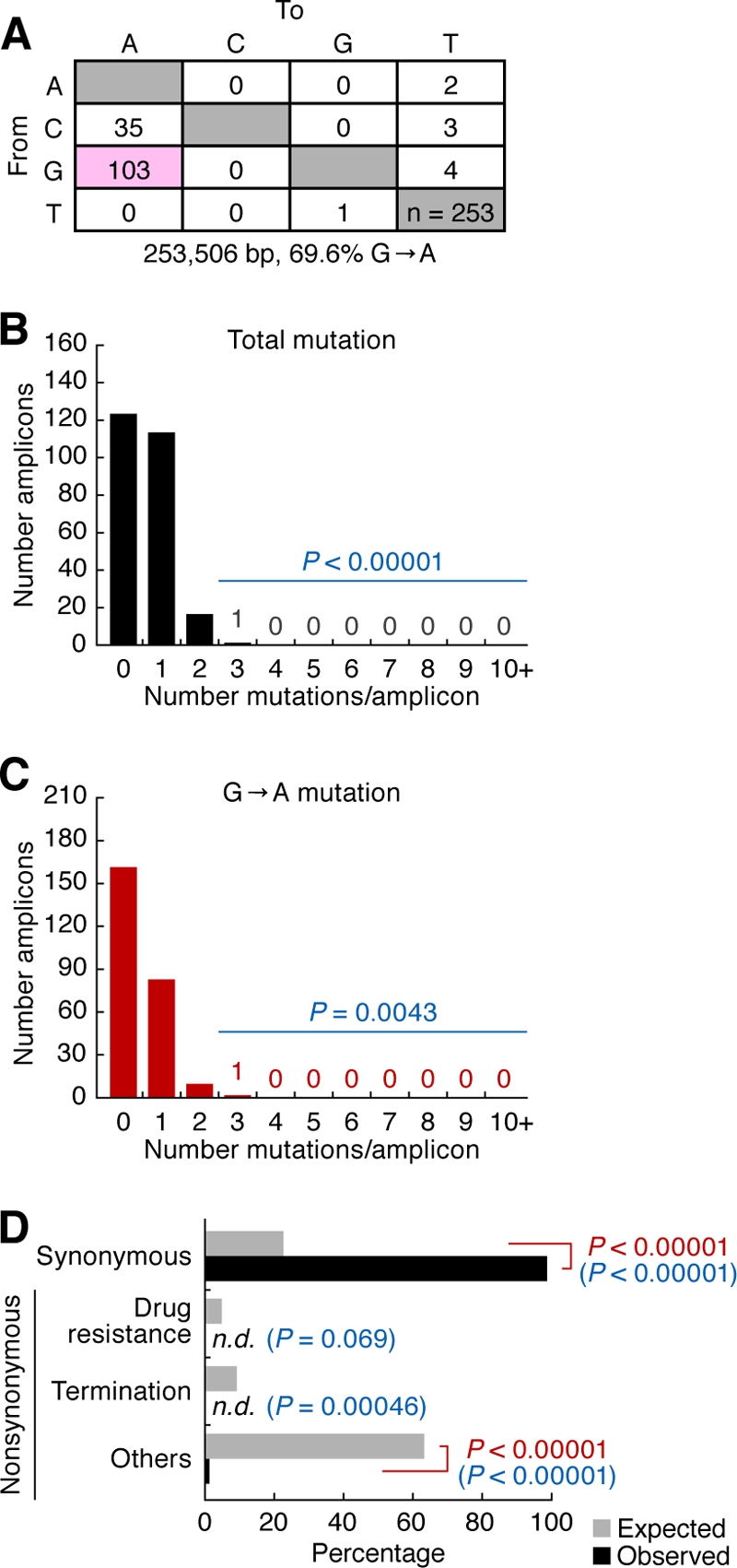
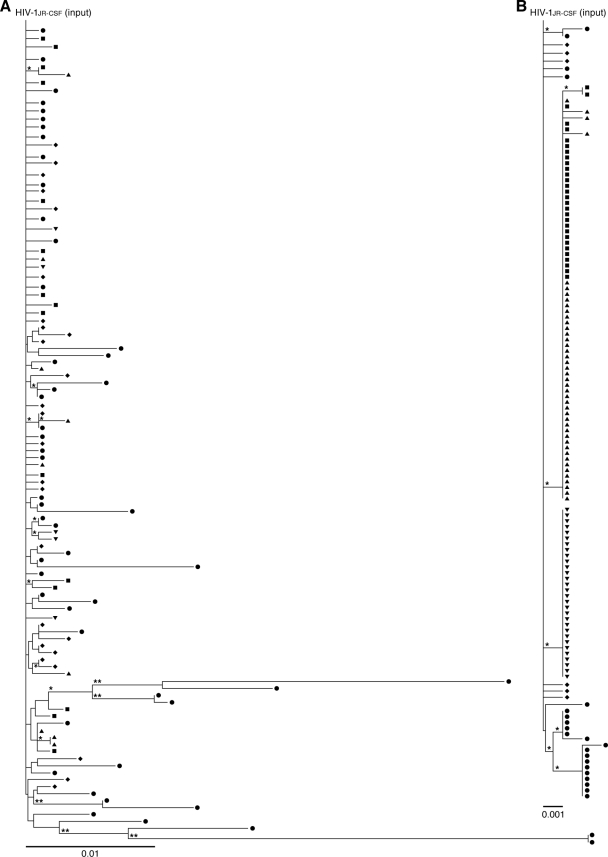
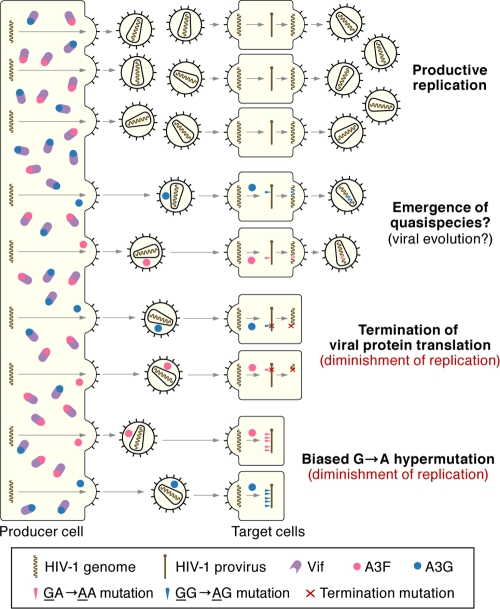
Genomic hypermutation of RNA viruses, including human immunodeficiency virus type 1 (HIV-1), can be provoked by intrinsic and extrinsic pressures, which lead to the inhibition of viral replication and/or the progression of viral diversity. Human APOBEC3G was identified as an HIV-1 restriction factor, which edits nascent HIV-1 DNA by inducing G-to-A hypermutations and debilitates the infectivity of vif-deficient HIV-1. On the other hand, HIV-1 Vif protein has the robust potential to degrade APOBEC3G protein. Although subsequent investigations have revealed that lines of APOBEC3 family proteins have the capacity to mutate HIV-1 DNA, it remains unclear whether these endogenous APOBEC3s, including APOBEC3G, contribute to mutations of vif-proficient HIV-1 provirus in vivo and, if so, what is the significance of these mutations. In this study, we use a human hematopoietic stem cell-transplanted humanized mouse (NOG-hCD34 mouse) model and demonstrate the predominant accumulation of G-to-A mutations in vif-proficient HIV-1 provirus displaying characteristics of APOBEC3-mediated mutagenesis. Notably, the APOBEC3-associated G-to-A mutation of HIV-1 DNA that leads to the termination of translation was significantly observed. We further provide a novel insight suggesting that HIV-1 G-to-A hypermutation is independently induced by individual APOBEC3 proteins. In contrast to the prominent mutation in intracellular proviral DNA, viral RNA in plasma possessed fewer G-to-A mutations. Taken together, these results provide the evidence indicating that endogenous APOBEC3s are associated with G-to-A mutation of HIV-1 provirus in vivo, which can result in the abrogation of HIV-1 infection.
Figures
Similar articles
-
Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n.PLoS Pathog. 2012;8(7):e1002800. doi: 10.1371/journal.ppat.1002800. Epub 2012 Jul 12. PLoS Pathog. 2012. PMID: 22807680 Free PMC article.
-
Simian Immunodeficiency Virus Vif and Human APOBEC3B Interactions Resemble Those between HIV-1 Vif and Human APOBEC3G.J Virol. 2018 May 29;92(12):e00447-18. doi: 10.1128/JVI.00447-18. Print 2018 Jun 15. J Virol. 2018. PMID: 29618650 Free PMC article.
-
APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis.J Virol. 2010 Jul;84(14):7396-404. doi: 10.1128/JVI.00056-10. Epub 2010 May 12. J Virol. 2010. PMID: 20463080 Free PMC article.
-
Running loose or getting lost: how HIV-1 counters and capitalizes on APOBEC3-induced mutagenesis through its Vif protein.Viruses. 2012 Nov 14;4(11):3132-61. doi: 10.3390/v4113132. Viruses. 2012. PMID: 23202519 Free PMC article. Review.
-
Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all.J Mol Biol. 2014 Mar 20;426(6):1220-45. doi: 10.1016/j.jmb.2013.10.033. Epub 2013 Nov 2. J Mol Biol. 2014. PMID: 24189052 Free PMC article. Review.
Cited by
-
New generation humanized mice for virus research: comparative aspects and future prospects.Virology. 2013 Jan 5;435(1):14-28. doi: 10.1016/j.virol.2012.10.007. Virology. 2013. PMID: 23217612 Free PMC article. Review.
-
Role of co-expressed APOBEC3F and APOBEC3G in inducing HIV-1 drug resistance.Heliyon. 2019 Apr 16;5(4):e01498. doi: 10.1016/j.heliyon.2019.e01498. eCollection 2019 Apr. Heliyon. 2019. PMID: 31025011 Free PMC article.
-
Experimental Adaptive Evolution of Simian Immunodeficiency Virus SIVcpz to Pandemic Human Immunodeficiency Virus Type 1 by Using a Humanized Mouse Model.J Virol. 2018 Jan 30;92(4):e01905-17. doi: 10.1128/JVI.01905-17. Print 2018 Feb 15. J Virol. 2018. PMID: 29212937 Free PMC article.
-
HIV-1 competition experiments in humanized mice show that APOBEC3H imposes selective pressure and promotes virus adaptation.PLoS Pathog. 2017 May 5;13(5):e1006348. doi: 10.1371/journal.ppat.1006348. eCollection 2017 May. PLoS Pathog. 2017. PMID: 28475648 Free PMC article.
-
Vpu augments the initial burst phase of HIV-1 propagation and downregulates BST2 and CD4 in humanized mice.J Virol. 2012 May;86(9):5000-13. doi: 10.1128/JVI.07062-11. Epub 2012 Feb 22. J Virol. 2012. PMID: 22357275 Free PMC article.
References
-
- Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S. J. Cho, and M. H. Malim. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14:1392-1396. - PubMed
-
- Chiu, Y. L., and W. C. Greene. 2008. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 26:317-353. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
