

Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy
- PMID: 20615414
- PMCID: PMC2954357
- DOI: 10.1016/j.yjmcc.2010.06.006
Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy
Abstract
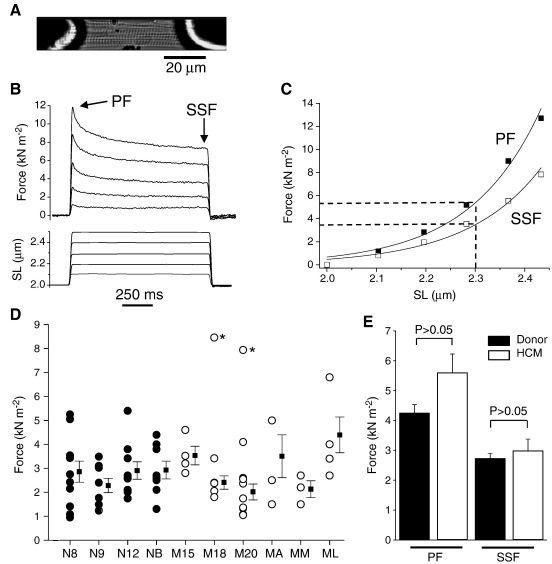
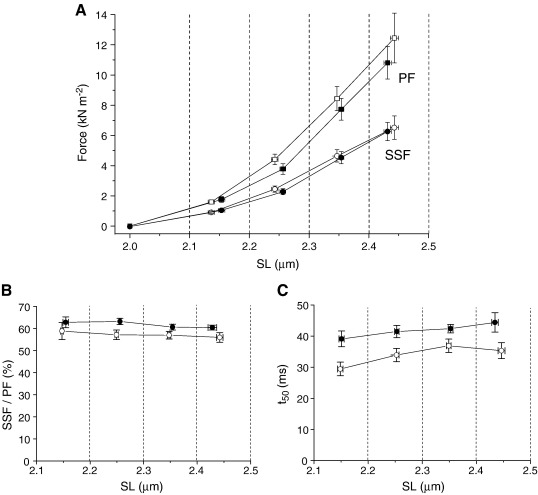
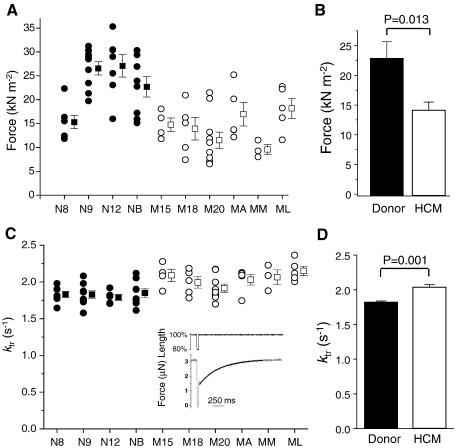
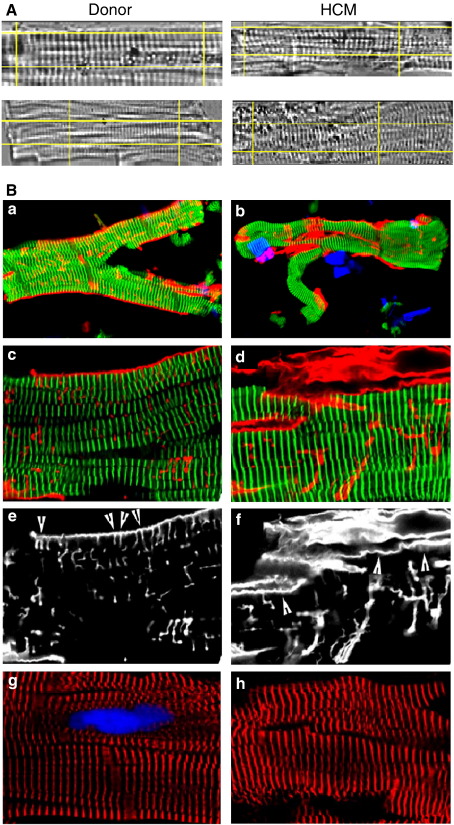
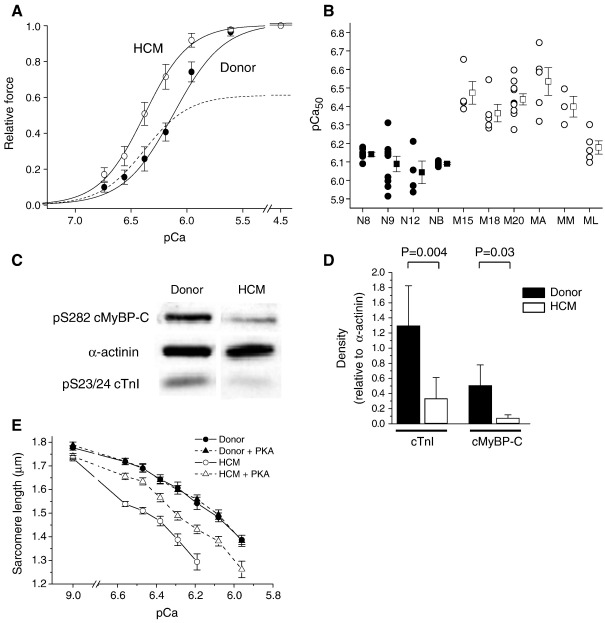
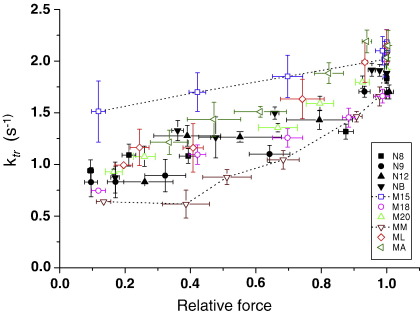
Hypertrophic cardiomyopathy (HCM) is characterized by left ventricular hypertrophy, increased ventricular stiffness and impaired diastolic filling. We investigated to what extent myocardial functional defects can be explained by alterations in the passive and active properties of human cardiac myofibrils. Skinned ventricular myocytes were prepared from patients with obstructive HCM (two patients with MYBPC3 mutations, one with a MYH7 mutation, and three with no mutation in either gene) and from four donors. Passive stiffness, viscous properties, and titin isoform expression were similar in HCM myocytes and donor myocytes. Maximal Ca(2+)-activated force was much lower in HCM myocytes (14 ± 1 kN/m(2)) than in donor myocytes (23 ± 3 kN/m(2); P<0.01), though cross-bridge kinetics (k(tr)) during maximal Ca(2)(+) activation were 10% faster in HCM myocytes. Myofibrillar Ca(2)(+) sensitivity in HCM myocytes (pCa(50)=6.40 ± 0.05) was higher than for donor myocytes (pCa(50)=6.09 ± 0.02; P<0.001) and was associated with reduced phosphorylation of troponin-I (ser-23/24) and MyBP-C (ser-282) in HCM myocytes. These characteristics were common to all six HCM patients and may therefore represent a secondary consequence of the known and unknown underlying genetic variants. Some HCM patients did however exhibit an altered relationship between force and cross-bridge kinetics at submaximal Ca(2+) concentrations, which may reflect the primary mutation. We conclude that the passive viscoelastic properties of the myocytes are unlikely to account for the increased stiffness of the HCM ventricle. However, the low maximum Ca(2+)-activated force and high Ca(2+) sensitivity of the myofilaments are likely to contribute substantially to any systolic and diastolic dysfunction, respectively, in hearts of HCM patients.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Figures
References
-
- Watkins H., Seidman J.G., Seidman C.E. Familial hypertrophic cardiomyopathy: a genetic model of cardiac hypertrophy. Hum Mol Genet. 1995;4:1721–1727. - PubMed
-
- Maron B.J. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. - PubMed
-
- Elliott P., McKenna W.J. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–1891. - PubMed
-
- Van Driest S.L., Vasile V.C., Ommen S.R., Will M.L., Tajik A.J., Gersh B.J. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
