

Intergenic and repeat transcription in human, chimpanzee and macaque brains measured by RNA-Seq
- PMID: 20617162
- PMCID: PMC2895644
- DOI: 10.1371/journal.pcbi.1000843
Intergenic and repeat transcription in human, chimpanzee and macaque brains measured by RNA-Seq
Abstract
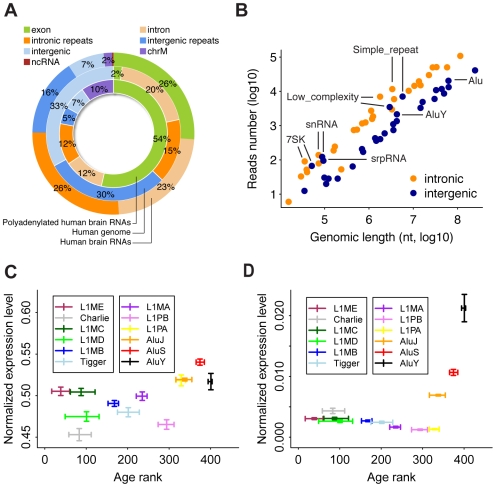
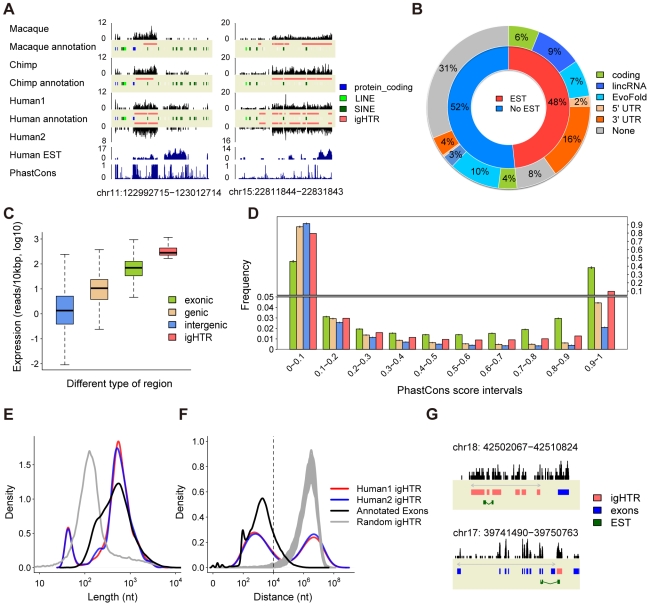
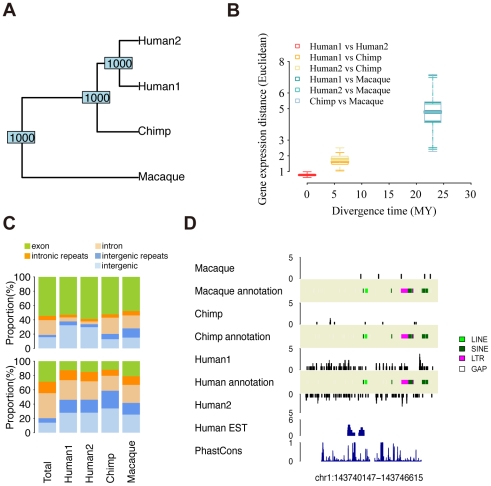
Transcription is the first step connecting genetic information with an organism's phenotype. While expression of annotated genes in the human brain has been characterized extensively, our knowledge about the scope and the conservation of transcripts located outside of the known genes' boundaries is limited. Here, we use high-throughput transcriptome sequencing (RNA-Seq) to characterize the total non-ribosomal transcriptome of human, chimpanzee, and rhesus macaque brain. In all species, only 20-28% of non-ribosomal transcripts correspond to annotated exons and 20-23% to introns. By contrast, transcripts originating within intronic and intergenic repetitive sequences constitute 40-48% of the total brain transcriptome. Notably, some repeat families show elevated transcription. In non-repetitive intergenic regions, we identify and characterize 1,093 distinct regions highly expressed in the human brain. These regions are conserved at the RNA expression level across primates studied and at the DNA sequence level across mammals. A large proportion of these transcripts (20%) represents 3'UTR extensions of known genes and may play roles in alternative microRNA-directed regulation. Finally, we show that while transcriptome divergence between species increases with evolutionary time, intergenic transcripts show more expression differences among species and exons show less. Our results show that many yet uncharacterized evolutionary conserved transcripts exist in the human brain. Some of these transcripts may play roles in transcriptional regulation and contribute to evolution of human-specific phenotypic traits.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- van Bakel H, Hughes TR. Establishing legitimacy and function in the new transcriptome. Brief Funct Genomic Proteomic. 2009;8:424–436. - PubMed
-
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–159. - PubMed
-
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. - PubMed
-
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
