

Osteopoikilosis and multiple exostoses caused by novel mutations in LEMD3 and EXT1 genes respectively--coincidence within one family
- PMID: 20618940
- PMCID: PMC2912259
- DOI: 10.1186/1471-2350-11-110
Osteopoikilosis and multiple exostoses caused by novel mutations in LEMD3 and EXT1 genes respectively--coincidence within one family
Abstract
Background: Osteopoikilosis is a rare autosomal dominant genetic disorder, characterised by the occurrence of the hyperostotic spots preferentially localized in the epiphyses and metaphyses of the long bones, and in the carpal and tarsal bones 1. Heterozygous LEMD3 gene mutations were shown to be the primary cause of the disease 2. Association of the primarily asymptomatic osteopokilosis with connective tissue nevi of the skin is categorized as Buschke-Ollendorff syndrome (BOS) 3. Additionally, osteopoikilosis can coincide with melorheostosis (MRO), a more severe bone disease characterised by the ectopic bone formation on the periosteal and endosteal surface of the long bones 456. However, not all MRO affected individuals carry germ-line LEMD3 mutations 7. Thus, the genetic cause of MRO remains unknown. Here we describe a familial case of osteopoikilosis in which a novel heterozygous LEMD3 mutation coincides with a novel mutation in EXT1, a gene involved in aetiology of multiple exostosis syndrome. The patients affected with both LEMD3 and EXT1 gene mutations displayed typical features of the osteopoikilosis. There were no additional skeletal manifestations detected however, various non-skeletal pathologies coincided in this group.
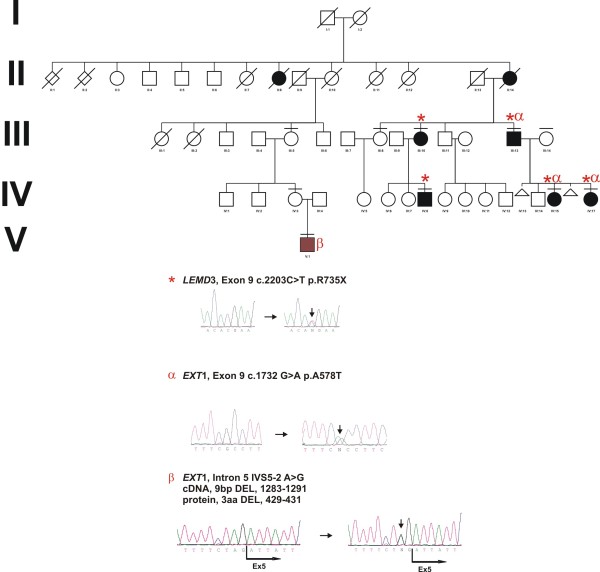
Methods: We investigated LEMD3 and EXT1 in the three-generation family from Poland, with 5 patients affected with osteopoikilosis and one child affected with multiple exostoses.
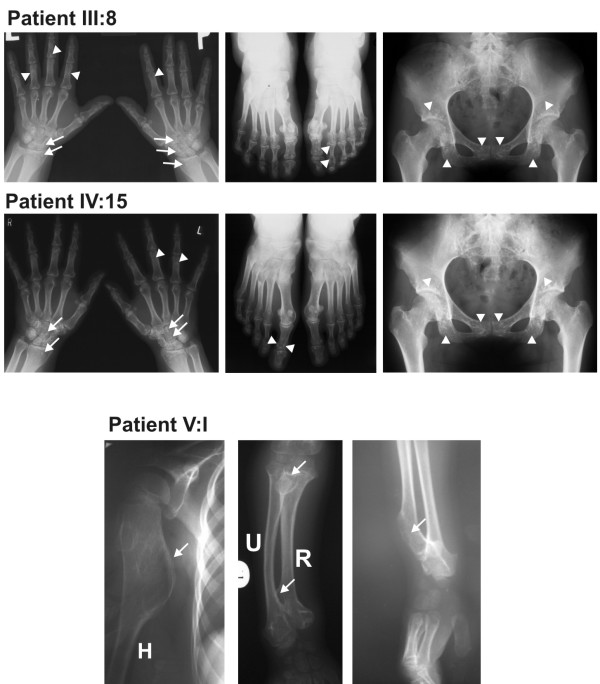
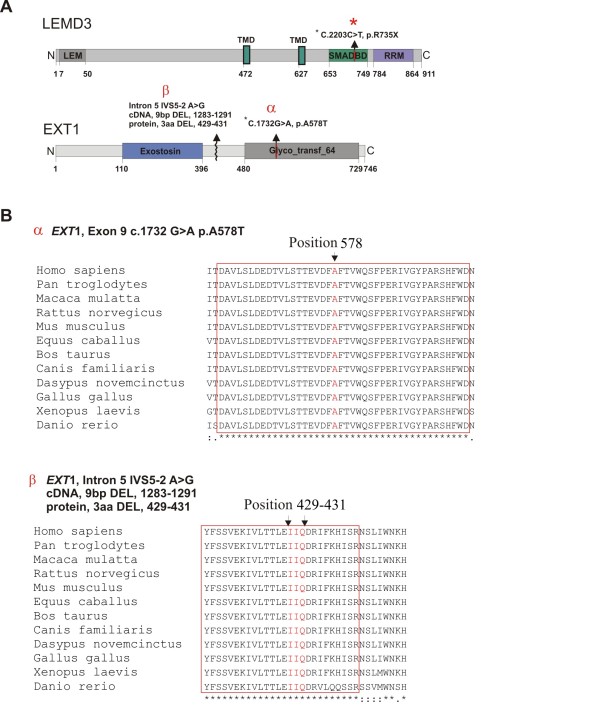
Results: We found a novel c.2203C > T (p.R735X) mutation in exon 9 of LEMD3, resulting in a premature stop codon at amino acid position 735. The mutation co-segregates with the osteopoikilosis phenotype and was not found in 200 ethnically matched controls. Another new substitution G > A was found in EXT1 gene at position 1732 (cDNA) in Exon 9 (p.A578T) in three out of five osteopoikilosis affected family members. Evolutionary conservation of the affected amino acid suggested possible functional relevance, however no additional skeletal manifestations were observed other then those specific for osteopoikilosis. Finally in one member of the family we found a splice site mutation in the EXT1 gene intron 5 (IVS5-2 A > G) resulting in the deletion of 9 bp of cDNA encoding three evolutionarily conserved amino acid residues. This child patient suffered from a severe form of exostoses, thus a causal relationship can be postulated.
Conclusions: We identified a new mutation in LEMD3 gene, accounting for the familial case of osteopoikilosis. In the same family we identified two novel EXT1 gene mutations. One of them A598T co-incided with the LEMD3 mutation. Co-incidence of LEMD3 and EXT1 gene mutations was not associated with a more severe skeletal phenotype in those patients.
Figures
References
-
- Melnick JC. Osteopathia condensans disseminata (osteopoikilosis); study of a family of 4 generations. Am J Roentgenol Radium Ther Nucl Med. 1959;82(2):229–238. - PubMed
-
- Hellemans J, Preobrazhenska O, Willaert A, Debeer P, Verdonk PC, Costa T, Janssens K, Menten B, Van Roy N, Vermeulen SJ. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet. 2004;36(11):1213–1218. doi: 10.1038/ng1453. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
