

EGF-induced Grb7 recruits and promotes Ras activity essential for the tumorigenicity of Sk-Br3 breast cancer cells
- PMID: 20622016
- PMCID: PMC2937960
- DOI: 10.1074/jbc.C110.114124
EGF-induced Grb7 recruits and promotes Ras activity essential for the tumorigenicity of Sk-Br3 breast cancer cells
Abstract
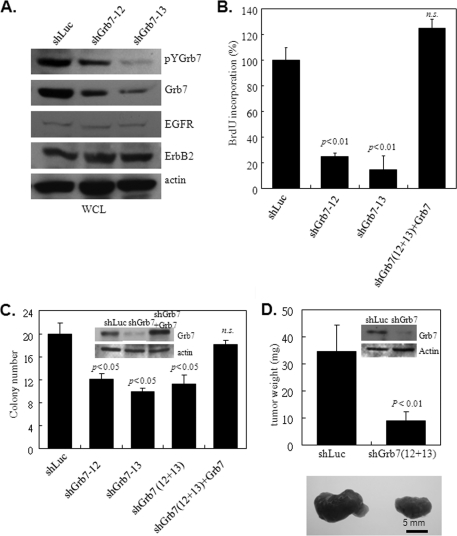
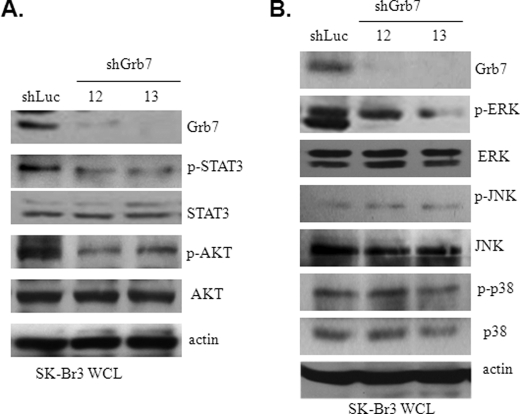
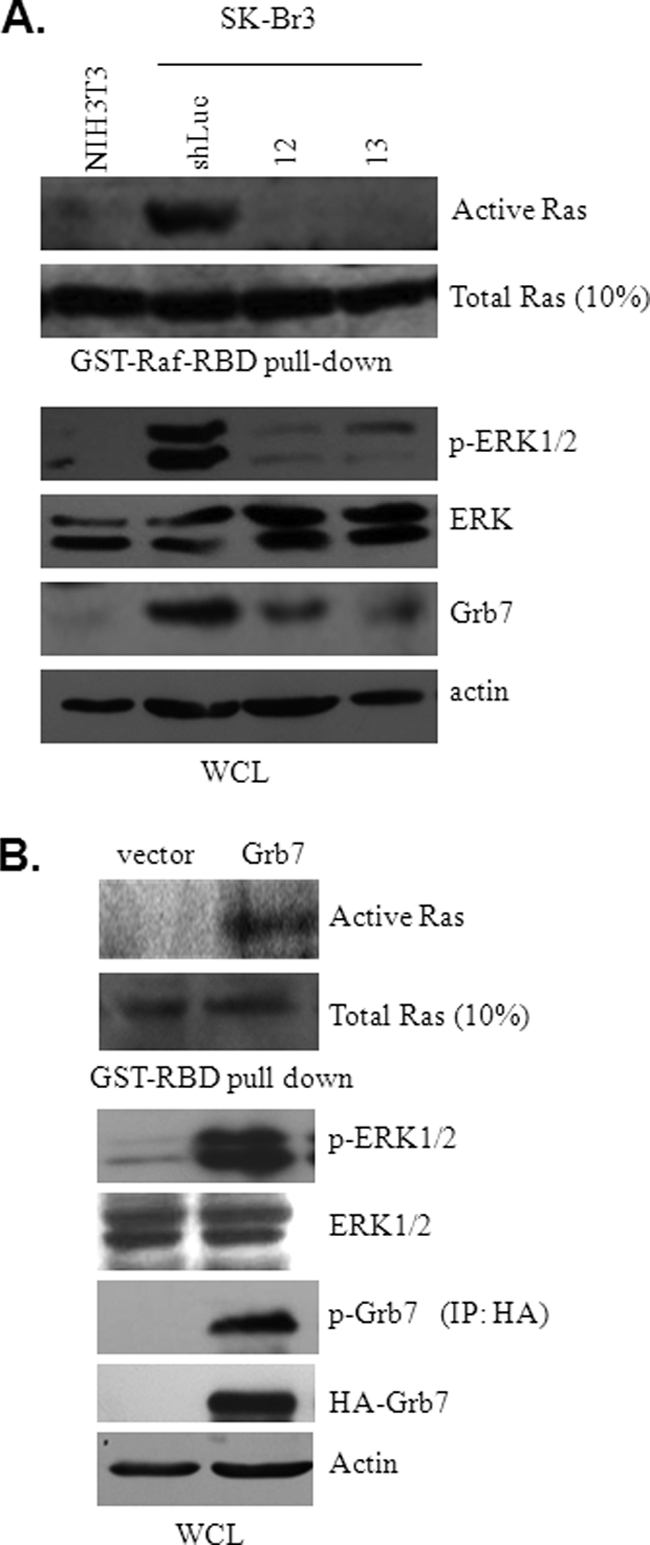
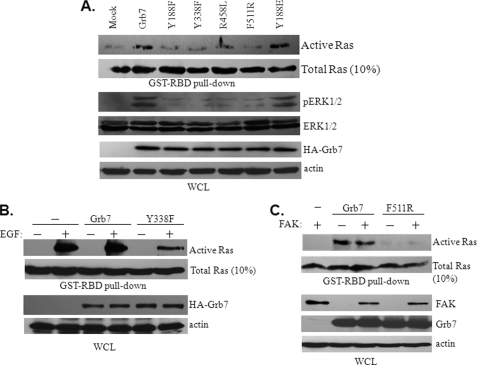
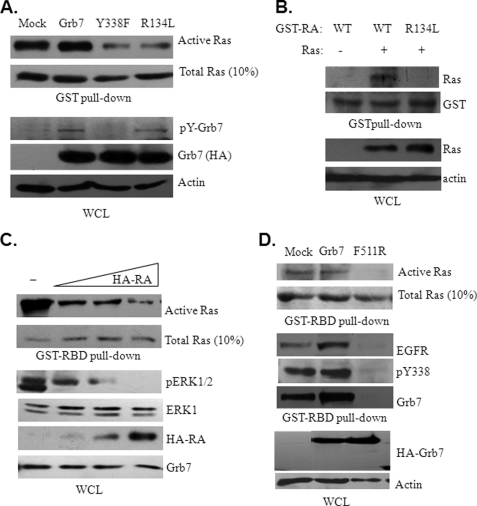
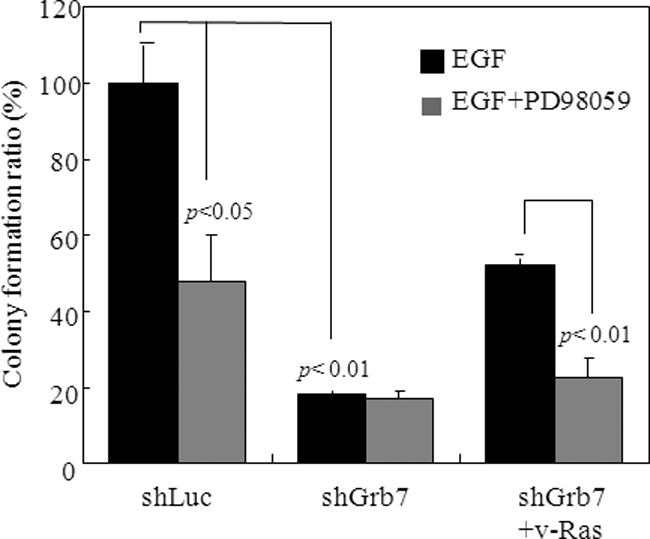
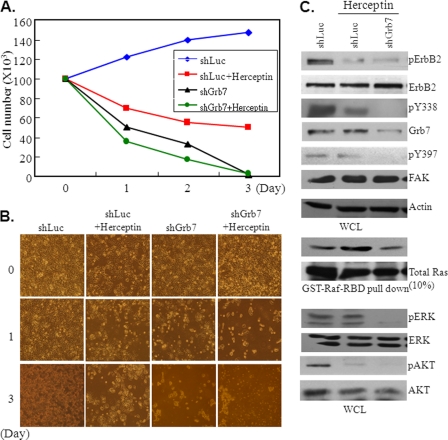
Co-amplification and co-overexpression of ErbB2 and Grb7 are frequently found in various cancers, including breast cancer. Biochemical and functional correlations of the two molecules have identified Grb7 to be a pivotal mediator downstream of ErbB2-mediated oncogenesis. However, it remains largely unknown how Grb7 is involve in the ErbB2-mediated tumorigenesis. In this study, we show that Grb7-mediated cell proliferation and growth are essential for the tumorigenesis that occurs in ErbB2-Grb7-overexpressing breast cancer cells. Intrinsically, EGF-induced de novo Grb7 tyrosine phosphorylation/activation recruits and activates Ras-GTPases and subsequently promotes the phosphorylation of ERK1/2, thereby stimulating tumor growth. Furthermore, we also found the anti-tumor effect could be synergized by co-treatment with Herceptin plus Grb7 knockdown in Sk-Br3 breast cancer cells. Our findings illustrate an underlying mechanism by which Grb7 promotes tumorigenesis through the formation of a novel EGFR-Grb7-Ras signaling complex, thereby highlighting the potential strategy of targeting Grb7 as an anti-breast cancer therapy.
Figures
References
-
- Han D. C., Shen T. L., Guan J. L. (2001) Oncogene 20, 6315–6321 - PubMed
-
- Shen T. L., Guan J. L. (2004) Front. Biosci. 9, 192–200 - PubMed
-
- Li H., Sánchez-Torres J., del Carpio A. F., Nogales-González A., Molina-Ortiz P., Moreno M. J., Török K., Villalobo A. (2005) Oncogene 24, 4206–4219 - PubMed
-
- Shen T. L., Han D. C., Guan J. L. (2002) J. Biol. Chem. 277, 29069–29077 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
