

HMGB1 release and redox regulates autophagy and apoptosis in cancer cells
- PMID: 20622903
- PMCID: PMC2945431
- DOI: 10.1038/onc.2010.261
HMGB1 release and redox regulates autophagy and apoptosis in cancer cells
Erratum in
-
Correction: HMGB1 release and redox regulates autophagy and apoptosis in cancer cells.Oncogene. 2025 Aug;44(32):2927-2928. doi: 10.1038/s41388-025-03510-0. Oncogene. 2025. PMID: 40707681 No abstract available.
Abstract
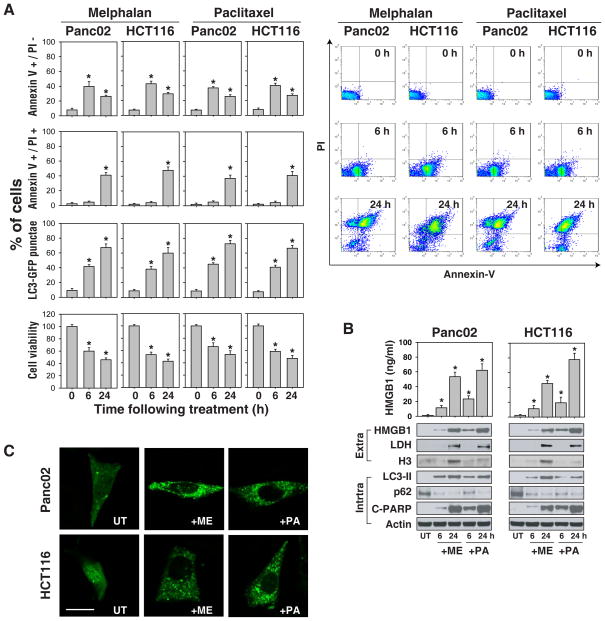
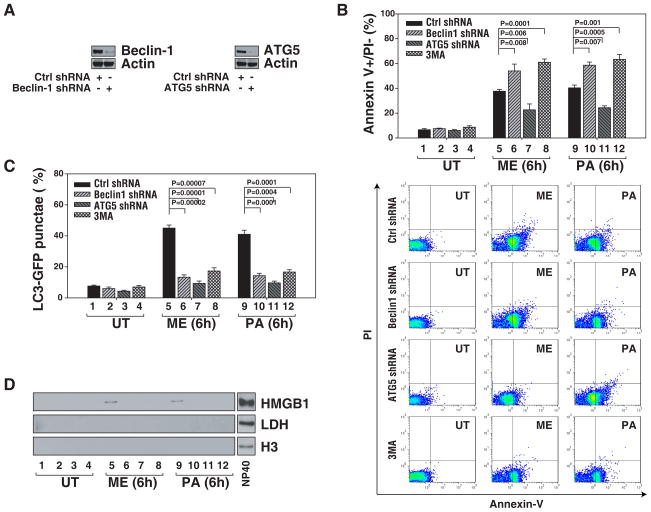
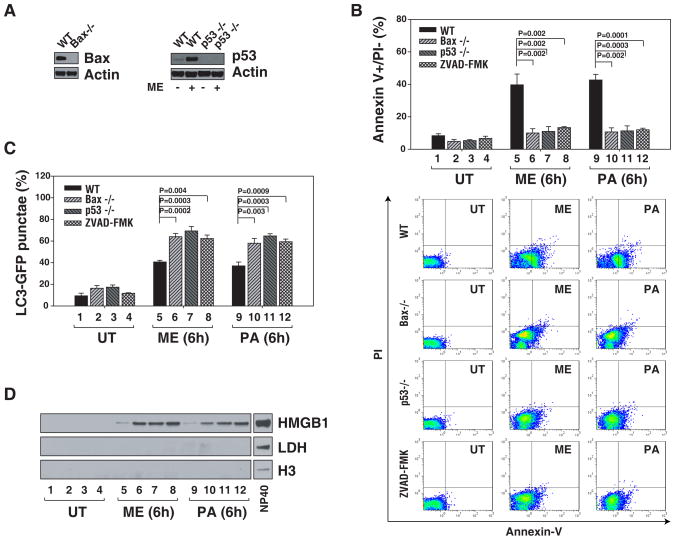
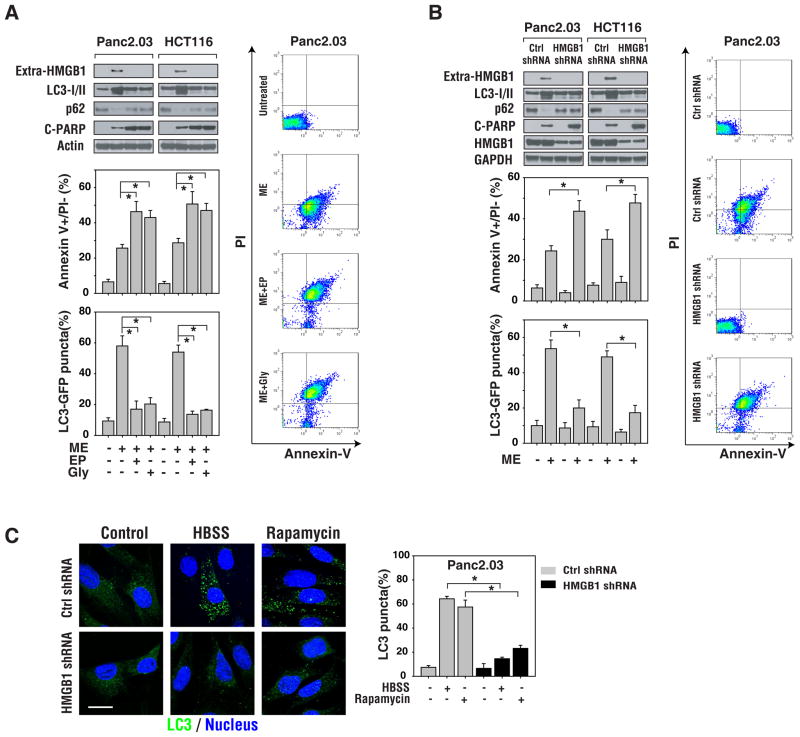
The functional relationship and cross-regulation between autophagy and apoptosis is complex. In this study we show that the high-mobility group box 1 protein (HMGB1) is a redox-sensitive regulator of the balance between autophagy and apoptosis. In cancer cells, anticancer agents enhanced autophagy and apoptosis, as well as HMGB1 release. HMGB1 release may be a prosurvival signal for residual cells after various cytotoxic cancer treatments. Diminished HMGB1 by short hairpin RNA transfection or inhibition of HMGB1 release by ethyl pyruvate or other small molecules led predominantly to apoptosis and decreased autophagy in stressed cancer cells. In this setting, reducible HMGB1 binds to the receptor for advanced glycation end products (RAGEs), but not to Toll-like receptor 4, induces Beclin1-dependent autophagy and promotes tumor resistance to alkylators (melphalan), tubulin disrupting agents (paclitaxel), DNA crosslinkers (ultraviolet light) and DNA intercalators (oxaliplatin or adriamycin). On the contrary, oxidized HMGB1 increases the cytotoxicity of these agents and induces apoptosis mediated by the caspase-9/-3 intrinsic pathway. HMGB1 release, as well as its redox state, thus links autophagy and apoptosis, representing a suitable target when coupled with conventional tumor treatments.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–9. - PubMed
-
- Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485–94. - PubMed
-
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9. - PubMed
-
- Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777–90. - PubMed
-
- Bell CW, Jiang W, Reich CF, 3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:C1318–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
