

Heme oxygenase in the regulation of vascular biology: from molecular mechanisms to therapeutic opportunities
- PMID: 20624029
- PMCID: PMC2988629
- DOI: 10.1089/ars.2010.3153
Heme oxygenase in the regulation of vascular biology: from molecular mechanisms to therapeutic opportunities
Abstract
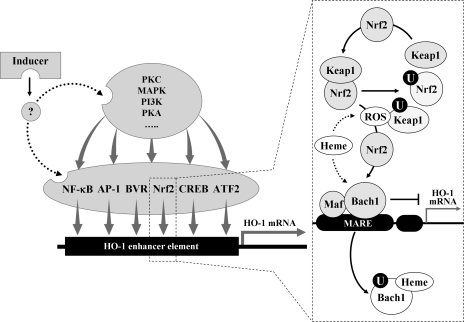
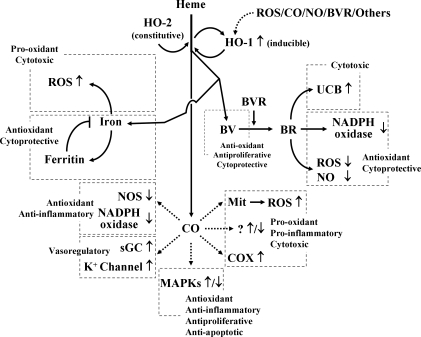
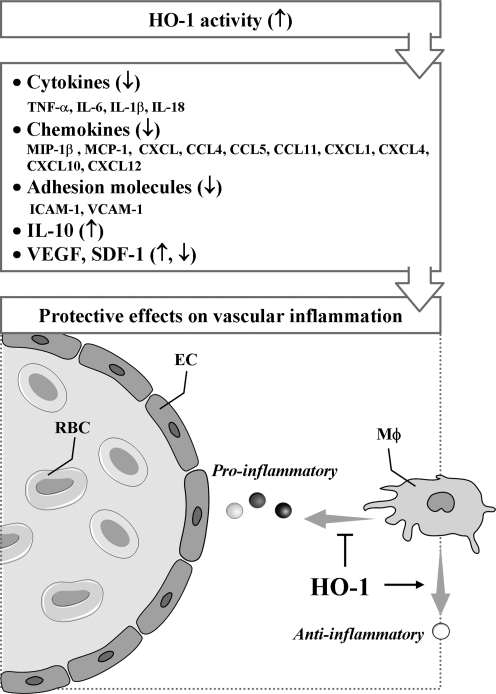
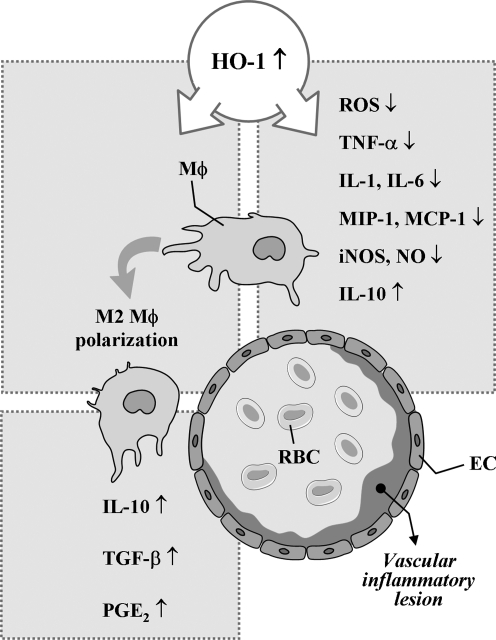
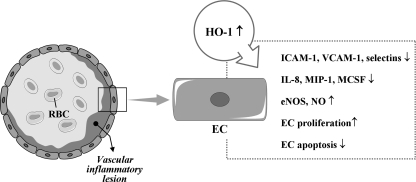
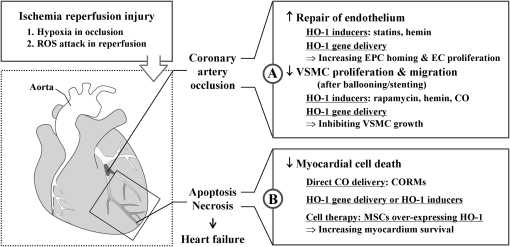
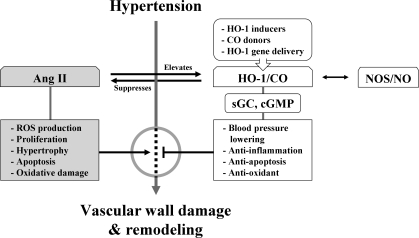
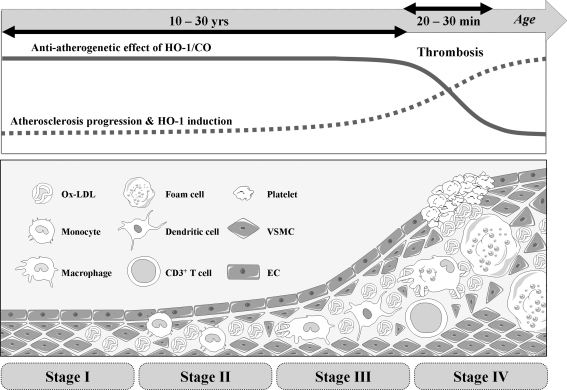
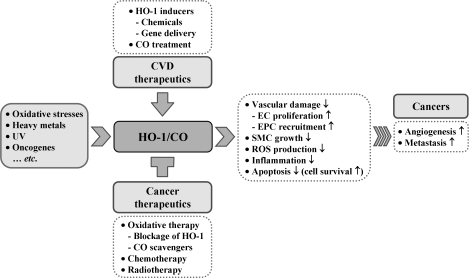
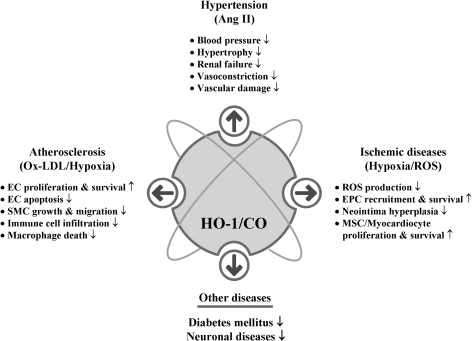
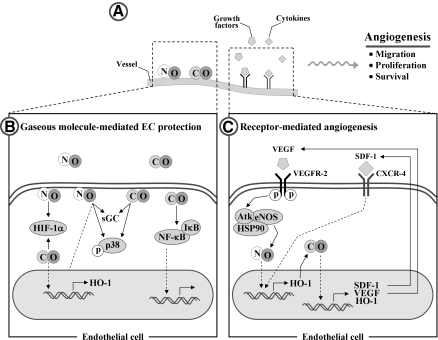
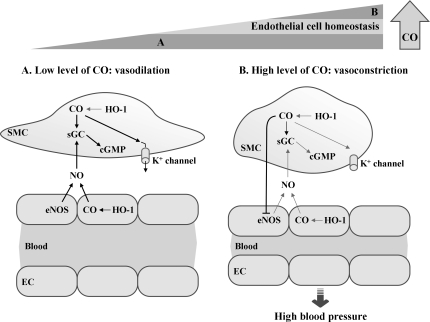
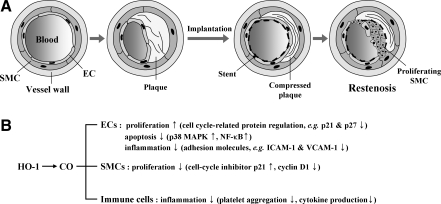
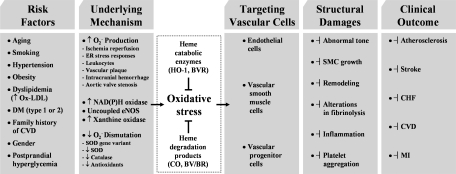
Heme oxygenases (HOs) are the rate-limiting enzymes in the catabolism of heme into biliverdin, free iron, and carbon monoxide. Two genetically distinct isoforms of HO have been characterized: an inducible form, HO-1, and a constitutively expressed form, HO-2. HO-1 is a kind of stress protein, and thus regarded as a sensitive and reliable indicator of cellular oxidative stress. The HO system acts as potent antioxidants, protects endothelial cells from apoptosis, is involved in regulating vascular tone, attenuates inflammatory response in the vessel wall, and participates in angiogenesis and vasculogenesis. Endothelial integrity and activity are thought to occupy the central position in the pathogenesis of cardiovascular diseases. Cardiovascular disease risk conditions converge in the contribution to oxidative stress. The oxidative stress leads to endothelial and vascular smooth muscle cell dysfunction with increases in vessel tone, cell growth, and gene expression that create a pro-thrombotic/pro-inflammatory environment. Subsequent formation, progression, and obstruction of atherosclerotic plaque may result in myocardial infarction, stroke, and cardiovascular death. This background provides the rationale for exploring the potential therapeutic role for HO system in the amelioration of vascular inflammation and prevention of adverse cardiovascular outcomes.
Figures

References
-
- Abraham NG. Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. - PubMed
-
- Abraham NG. Kushida T. McClung J. Weiss M. Quan S. Lafaro R. Darzynkiewicz Z. Wolin M. Heme oxygenase-1 attenuates glucose-mediated cell growth arrest and apoptosis in human microvessel endothelial cells. Circ Res. 2003;93:507–514. - PubMed
-
- Abraham NG. Rezzani R. Rodella L. Kruger A. Taller D. Li Volti G. Goodman AI. Kappas A. Overexpression of human heme oxygenase-1 attenuates endothelial cell sloughing in experimental diabetes. Am J Physiol Heart Circ Physiol. 2004;287:H2468–H2477. - PubMed
-
- Abraham NG. Tsenovoy PL. McClung J. Drummond GS. Heme oxygenase: a target gene for anti-diabetic and obesity. Curr Pharm Des. 2008;14:412–421. - PubMed
-
- Abuarqoub H. Foresti R. Green CJ. Motterlini R. Heme oxygenase-1 mediates the anti-inflammatory actions of 2'-hydroxychalcone in RAW 264.7 murine macrophages. Am J Physiol Cell Physiol. 2006;290:C1092–C1099. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical