

Deep genomic-scale analyses of the metazoa reject Coelomata: evidence from single- and multigene families analyzed under a supertree and supermatrix paradigm
- PMID: 20624736
- PMCID: PMC2997542
- DOI: 10.1093/gbe/evq016
Deep genomic-scale analyses of the metazoa reject Coelomata: evidence from single- and multigene families analyzed under a supertree and supermatrix paradigm
Abstract
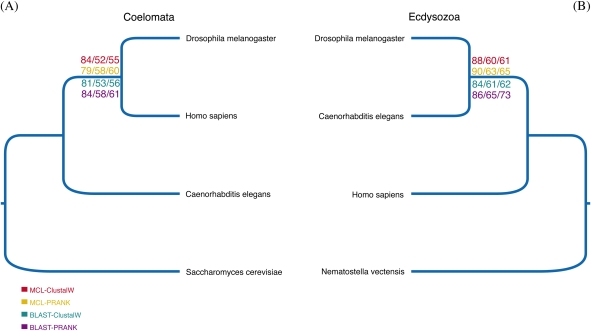
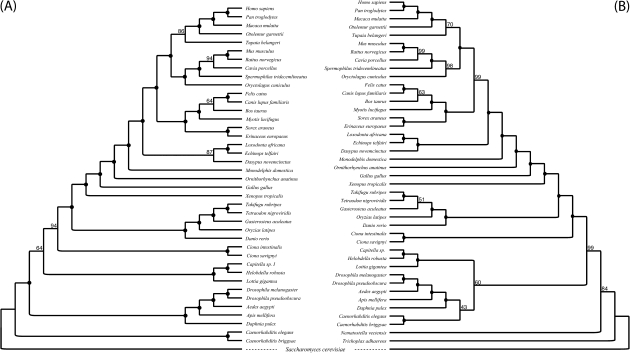
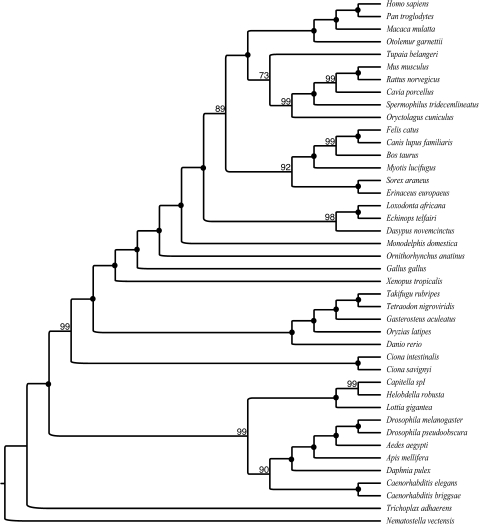
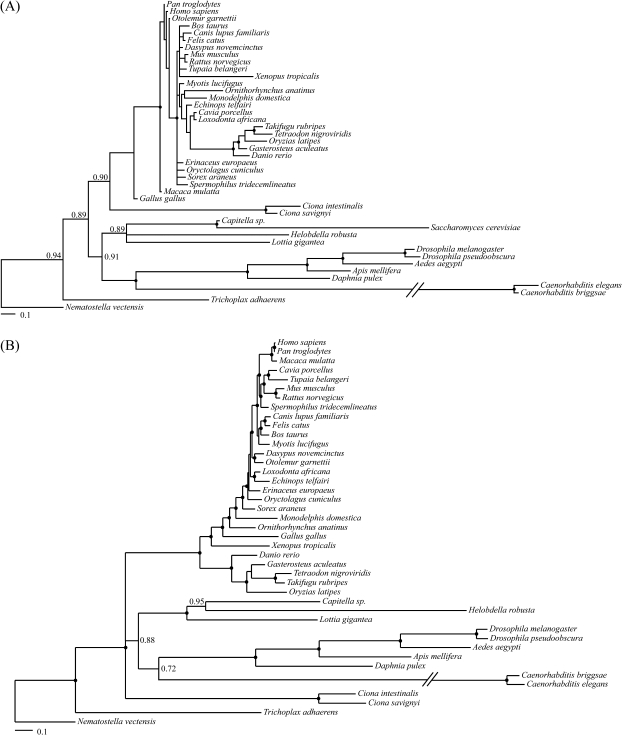
Solving the phylogeny of the animals with bilateral symmetry has proven difficult. Morphological studies have suggested a variety of alternative hypotheses, of which, Hyman's Coelomata hypothesis has become the most established. Studies based on 18S rRNA have failed to endorse Coelomata, supporting instead the rearrangement of the protostomes into two new clades: the Lophotrochozoa (including, e.g., the molluscs and the annelids) and the Ecdysozoa (including the Panarthropoda and most pseudocoelomates, such as the nematodes and priapulids). Support for this new animal phylogeny has been attained from expressed sequence tag studies, although these generally have a limited gene sampling. In contrast, deep genomic-scale analyses have often supported Coelomata. However, these studies are problematic due to their limited taxonomic sampling, which could exacerbate tree reconstruction artifacts. Here, we address both of these sampling limitations; we study the effect of long-branch attraction (LBA) in deep genomic-scale analyses and provide convincing evidence, using both single- and multigene families, that Coelomata is an artifact. We show that optimal outgroup selection is key in avoiding LBA and identify the use of inadequate outgroups as the reason previous deep genomic-scale analyses found strong support for Coelomata.
Figures
References
-
- Aguinaldo AMA, et al. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature. 1997;387:489–493. - PubMed
-
- Archie JW. A randomization test for phylogenetic information in systematic data. Syst Zool. 1989;38:219–225.
-
- Baum BR. Combining trees as a way of combining data sets for phylogenetic inference, and the desirability of combining gene trees. Taxon. 1992;41:3–10.
-
- Belinky F, Cohen O, Huchon D. Large-scale parsimony analysis of metazoan indels in protein-coding genes. Mol Biol Evol. 2010;27:441–451. - PubMed
