

A multilevel model to address batch effects in copy number estimation using SNP arrays
- PMID: 20625178
- PMCID: PMC3006124
- DOI: 10.1093/biostatistics/kxq043
A multilevel model to address batch effects in copy number estimation using SNP arrays
Abstract
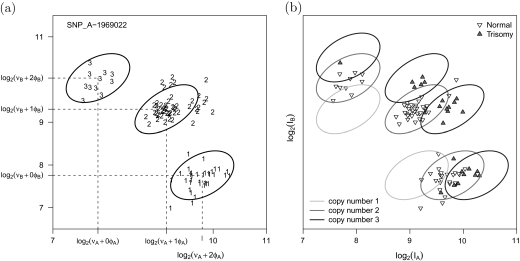
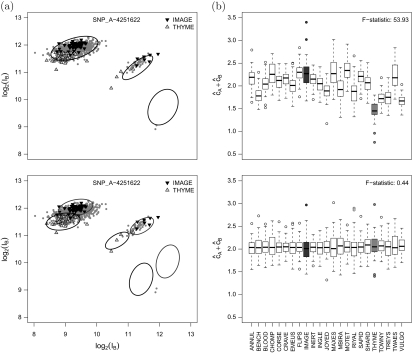
Submicroscopic changes in chromosomal DNA copy number dosage are common and have been implicated in many heritable diseases and cancers. Recent high-throughput technologies have a resolution that permits the detection of segmental changes in DNA copy number that span thousands of base pairs in the genome. Genomewide association studies (GWAS) may simultaneously screen for copy number phenotype and single nucleotide polymorphism (SNP) phenotype associations as part of the analytic strategy. However, genomewide array analyses are particularly susceptible to batch effects as the logistics of preparing DNA and processing thousands of arrays often involves multiple laboratories and technicians, or changes over calendar time to the reagents and laboratory equipment. Failure to adjust for batch effects can lead to incorrect inference and requires inefficient post hoc quality control procedures to exclude regions that are associated with batch. Our work extends previous model-based approaches for copy number estimation by explicitly modeling batch and using shrinkage to improve locus-specific estimates of copy number uncertainty. Key features of this approach include the use of biallelic genotype calls from experimental data to estimate batch-specific and locus-specific parameters of background and signal without the requirement of training data. We illustrate these ideas using a study of bipolar disease and a study of chromosome 21 trisomy. The former has batch effects that dominate much of the observed variation in the quantile-normalized intensities, while the latter illustrates the robustness of our approach to a data set in which approximately 27% of the samples have altered copy number. Locus-specific estimates of copy number can be plotted on the copy number scale to investigate mosaicism and guide the choice of appropriate downstream approaches for smoothing the copy number as a function of physical position. The software is open source and implemented in the R package crlmm at Bioconductor (http:www.bioconductor.org).
Figures


References
-
- Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S others. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:20007–20012. - PMC - PubMed
-
- Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, Grammastro MD, Sciarrotta MG, Buttitta F, Incarbone M others. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. Journal of Clinical Oncology. 2009;27:1667–1674. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
