

MER41 repeat sequences contain inducible STAT1 binding sites
- PMID: 20625510
- PMCID: PMC2897888
- DOI: 10.1371/journal.pone.0011425
MER41 repeat sequences contain inducible STAT1 binding sites
Abstract
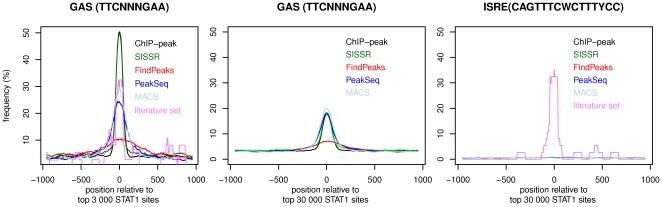
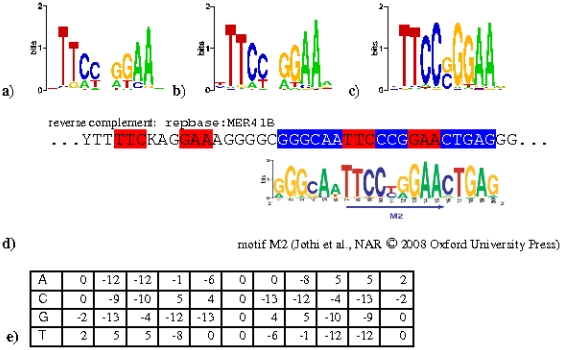
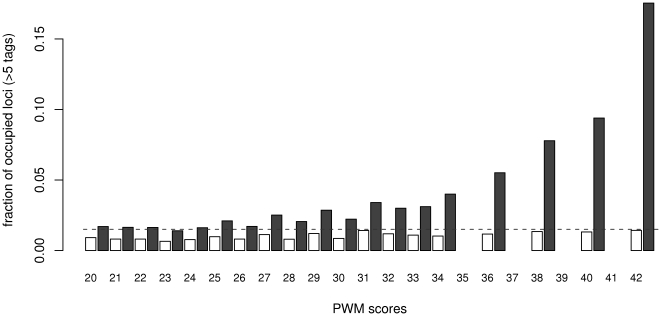
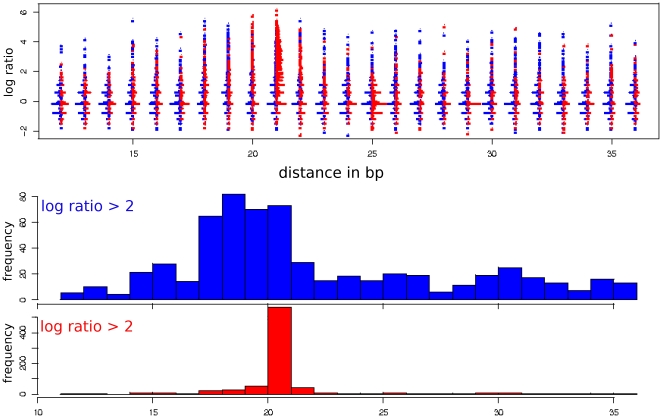
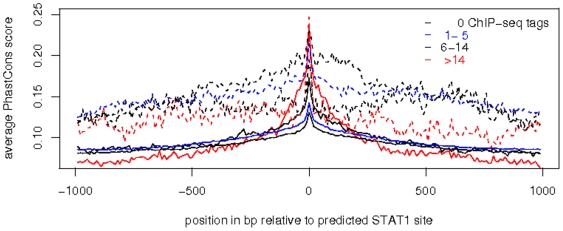
Chromatin immunoprecipitation combined with massively parallel sequencing methods (ChIP-seq) is becoming the standard approach to study interactions of transcription factors (TF) with genomic sequences. At the example of public STAT1 ChIP-seq data sets, we present novel approaches for the interpretation of ChIP-seq data.We compare recently developed approaches to determine STAT1 binding sites from ChIP-seq data. Assessing the content of the established consensus sequence for STAT1 binding sites, we find that the usage of "negative control" ChIP-seq data fails to provide substantial advantages. We derive a single refined probabilistic model of STAT1 binding sequences from these ChIP-seq data. Contrary to previous claims, we find no evidence that STAT1 binds to multiple distinct motifs upon interferon-gamma stimulation in vivo. While a large majority of genomic sites with high ChIP-seq signal is associated with a nucleotide sequence resembling a STAT1 binding site, only a very small subset of the over 5 million potential STAT1 binding sites in the human genome is covered by ChIP-seq data. Furthermore a surprisingly large fraction of the ChIP-seq signal (5%) is absorbed by a small family of repetitive sequences (MER41). The observation of the binding of activated STAT1 protein to a specific repetitive element bolsters similar reports concerning p53 and other TFs, and strengthens the notion of an involvement of repeats in gene regulation. Incidentally MER41 are specific to primates, consequently, regulatory mechanisms in the IFN-STAT pathway might fundamentally differ between primates and rodents. On a methodological aspect, the presence of large numbers of nearly identical binding sites in repetitive sequences may lead to wrong conclusions about intrinsic binding preferences of TF as illustrated by the spacing analysis STAT1 tandem motifs. Therefore, ChIP-seq data should be analyzed independently within repetitive and non-repetitive sequences.
Conflict of interest statement
Figures
References
-
- Tompa M, Li N, Bailey TL, Church GM, De Moor B, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005;23:137–144. - PubMed
-
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. - PubMed
-
- Schmid CD, Bucher P. ChIP-Seq Data Reveal Nucleosome Architecture of Human Promoters. Cell. 2007;131:831–832. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
