

BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments
- PMID: 20626897
- PMCID: PMC3017758
- DOI: 10.1186/1471-2148-10-210
BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments
Abstract
Background: The quality of multiple sequence alignments plays an important role in the accuracy of phylogenetic inference. It has been shown that removing ambiguously aligned regions, but also other sources of bias such as highly variable (saturated) characters, can improve the overall performance of many phylogenetic reconstruction methods. A current scientific trend is to build phylogenetic trees from a large number of sequence datasets (semi-)automatically extracted from numerous complete genomes. Because these approaches do not allow a precise manual curation of each dataset, there exists a real need for efficient bioinformatic tools dedicated to this alignment character trimming step.
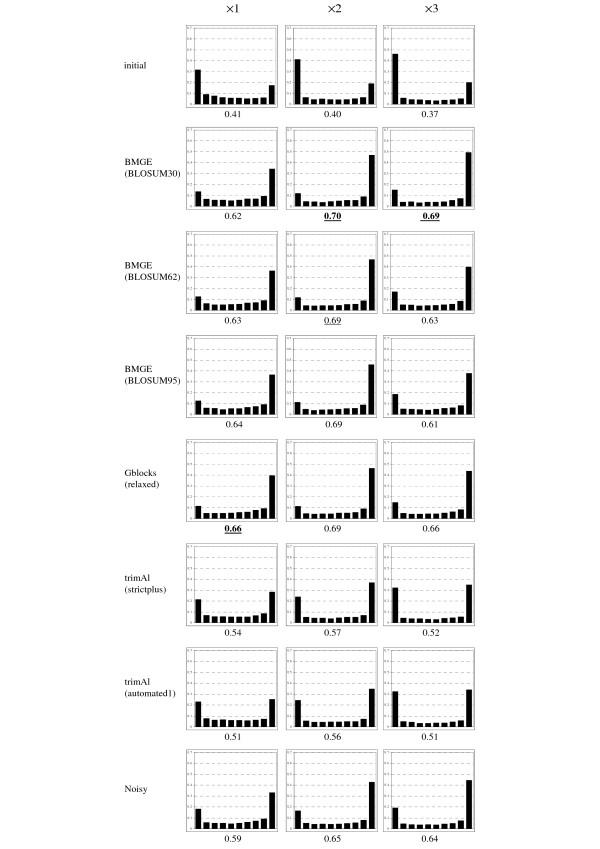
Results: Here is presented a new software, named BMGE (Block Mapping and Gathering with Entropy), that is designed to select regions in a multiple sequence alignment that are suited for phylogenetic inference. For each character, BMGE computes a score closely related to an entropy value. Calculation of these entropy-like scores is weighted with BLOSUM or PAM similarity matrices in order to distinguish among biologically expected and unexpected variability for each aligned character. Sets of contiguous characters with a score above a given threshold are considered as not suited for phylogenetic inference and then removed. Simulation analyses show that the character trimming performed by BMGE produces datasets leading to accurate trees, especially with alignments including distantly-related sequences. BMGE also implements trimming and recoding methods aimed at minimizing phylogeny reconstruction artefacts due to compositional heterogeneity.
Conclusions: BMGE is able to perform biologically relevant trimming on a multiple alignment of DNA, codon or amino acid sequences. Java source code and executable are freely available at ftp://ftp.pasteur.fr/pub/GenSoft/projects/BMGE/.
Figures







References
-
- Lake JA. The order of sequence alignment can bias the selection of tree topology. Mol Biol Evol. 1991;8:378–385. - PubMed
-
- Morrison DA, Ellis JT. Effects of nucleotide sequence alignment on phylogeny estimation: a case study on 18 S rDNAs of apicomplexa. Mol Biol Evol. 1997;14:428–441. - PubMed
-
- Wang L-S, Leebens-Mack J, Wall PK, Beckmann K, dePamphilis CW, Warnow T. The impact of multiple protein sequence alignment on phylogenetic estimation. IEEE/ACM Trans Comput Biol Bioinf. 2009. in press . - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
