

Pressurized pepsin digestion in proteomics: an automatable alternative to trypsin for integrated top-down bottom-up proteomics
- PMID: 20627868
- PMCID: PMC3033671
- DOI: 10.1074/mcp.M110.001479
Pressurized pepsin digestion in proteomics: an automatable alternative to trypsin for integrated top-down bottom-up proteomics
Abstract
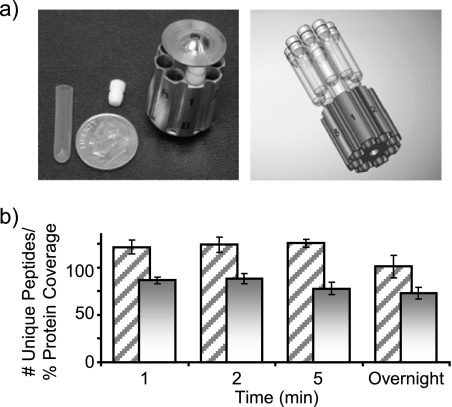
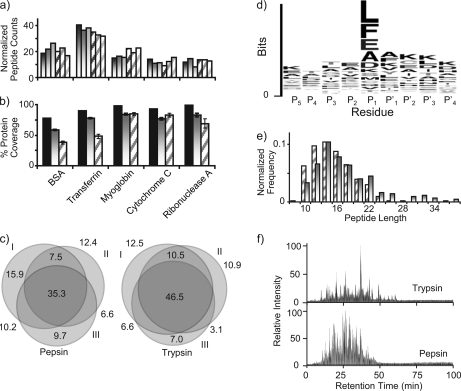
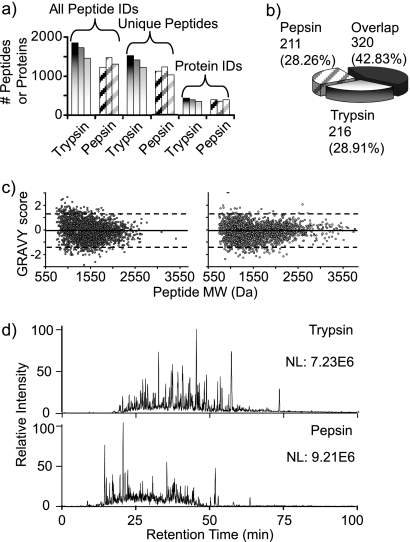
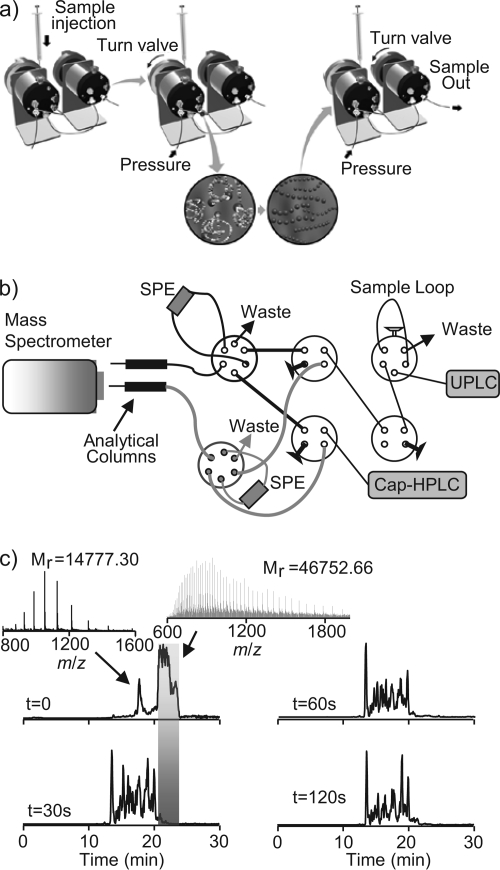
Integrated top-down bottom-up proteomics combined with on-line digestion has great potential to improve the characterization of protein isoforms in biological systems and is amendable to high throughput proteomics experiments. Bottom-up proteomics ultimately provides the peptide sequences derived from the tandem MS analyses of peptides after the proteome has been digested. Top-down proteomics conversely entails the MS analyses of intact proteins for more effective characterization of genetic variations and/or post-translational modifications. Herein, we describe recent efforts toward efficient integration of bottom-up and top-down LC-MS-based proteomics strategies. Since most proteomics separations utilize acidic conditions, we exploited the compatibility of pepsin (where the optimal digestion conditions are at low pH) for integration into bottom-up and top-down proteomics work flows. Pressure-enhanced pepsin digestions were successfully performed and characterized with several standard proteins in either an off-line mode using a Barocycler or an on-line mode using a modified high pressure LC system referred to as a fast on-line digestion system (FOLDS). FOLDS was tested using pepsin and a whole microbial proteome, and the results were compared against traditional trypsin digestions on the same platform. Additionally, FOLDS was integrated with a RePlay configuration to demonstrate an ultrarapid integrated bottom-up top-down proteomics strategy using a standard mixture of proteins and a monkey pox virus proteome.
Figures
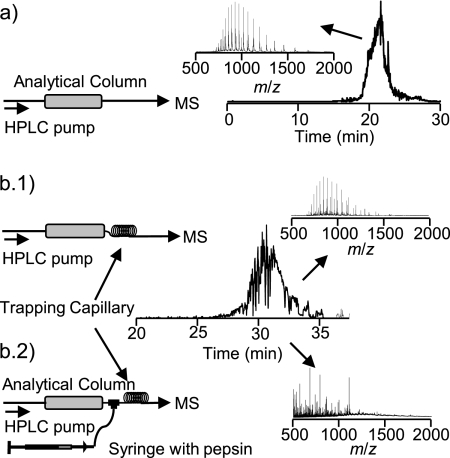
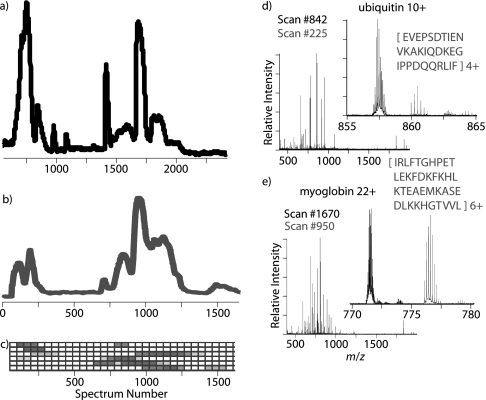
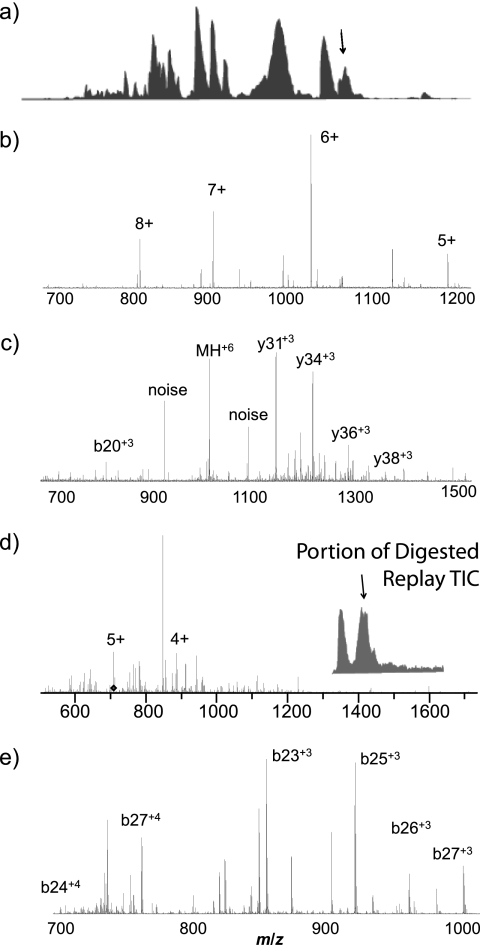
References
-
- Kelleher N. L., Taylor S. V., Grannis D., Kinsland C., Chiu H. J., Begley T. P., McLafferty F. W. (1998) Efficient sequence analysis of the six gene products (7–74 kDa) from the Escherichia coli thiamin biosynthetic operon by tandem high-resolution mass spectrometry. Protein Sci. 7, 1796–1801 - PMC - PubMed
-
- Zubarev R. A., Horn D. M., Fridriksson E. K., Kelleher N. L., Kruger N. A., Lewis M. A., Carpenter B. K., McLafferty F. W. (2000) Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 72, 563–573 - PubMed
-
- Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 - PubMed
-
- Gevaert K., Goethals M., Martens L., Van Damme J., Staes A., Thomas G. R., Vandekerckhove J. (2003) Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides. Nat. Biotechnol. 21, 566–569 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
