

Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: delineation of conserved core and lineage-specific transcriptomes during intracellular survival
- PMID: 20628579
- PMCID: PMC2900310
- DOI: 10.1371/journal.ppat.1000988
Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: delineation of conserved core and lineage-specific transcriptomes during intracellular survival
Abstract
Tuberculosis exerts a tremendous burden on global health, with approximately 9 million new infections and approximately 2 million deaths annually. The Mycobacterium tuberculosis complex (MTC) was initially regarded as a highly homogeneous population; however, recent data suggest the causative agents of tuberculosis are more genetically and functionally diverse than appreciated previously. The impact of this natural variation on the virulence and clinical manifestations of the pathogen remains largely unknown. This report examines the effect of genetic diversity among MTC clinical isolates on global gene expression and survival within macrophages. We discovered lineage-specific transcription patterns in vitro and distinct intracellular growth profiles associated with specific responses to host-derived environmental cues. Strain comparisons also facilitated delineation of a core intracellular transcriptome, including genes with highly conserved regulation across the global panel of clinical isolates. This study affords new insights into the genetic information that M. tuberculosis has conserved under selective pressure during its long-term interactions with its human host.
Conflict of interest statement
The authors have declared that no competing interests exist.
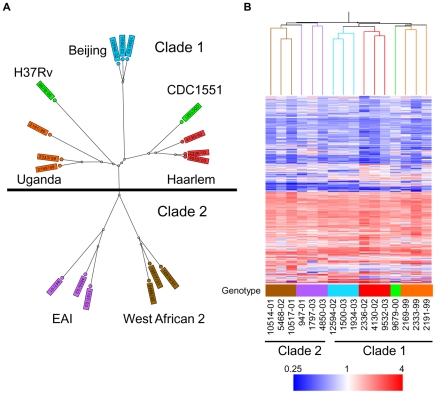
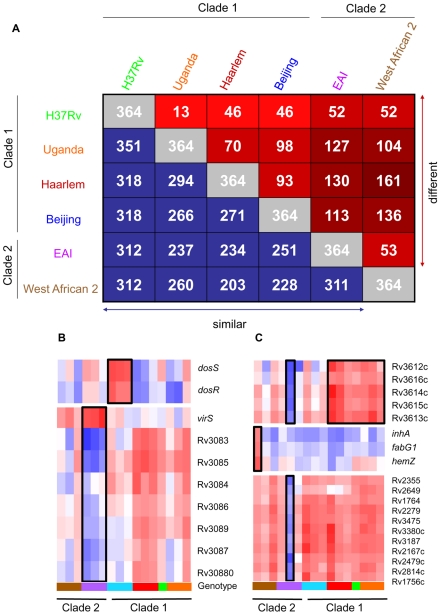
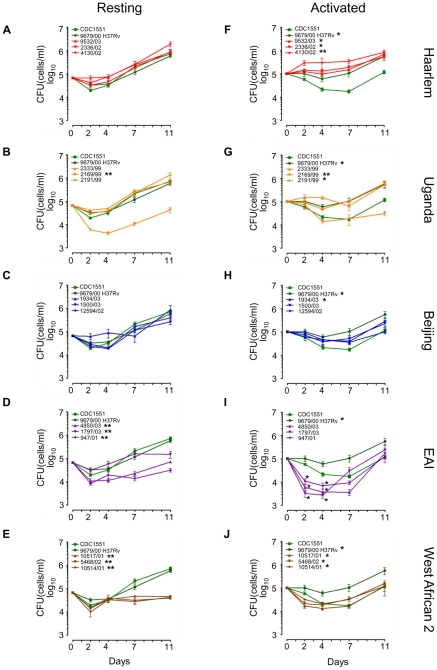
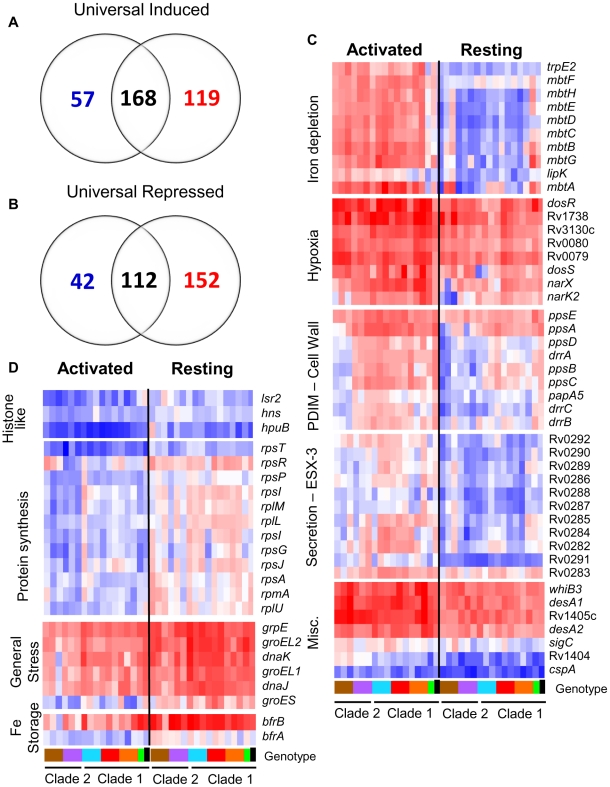
Figures
References
-
- Sousa AO, Salem JI, Lee FK, Verçosa MC, Cruaud P, et al. An epidemic of tuberculosis with a high rate of tuberculin anergy among a population previously unexposed to tuberculosis, the Yanomami Indians of the Brazilian Amazon. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13227–13232. - PMC - PubMed
-
- Achtman M. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol. 2008;62:53–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
