

MetaPIGA v2.0: maximum likelihood large phylogeny estimation using the metapopulation genetic algorithm and other stochastic heuristics
- PMID: 20633263
- PMCID: PMC2912891
- DOI: 10.1186/1471-2105-11-379
MetaPIGA v2.0: maximum likelihood large phylogeny estimation using the metapopulation genetic algorithm and other stochastic heuristics
Abstract
Background: The development, in the last decade, of stochastic heuristics implemented in robust application softwares has made large phylogeny inference a key step in most comparative studies involving molecular sequences. Still, the choice of a phylogeny inference software is often dictated by a combination of parameters not related to the raw performance of the implemented algorithm(s) but rather by practical issues such as ergonomics and/or the availability of specific functionalities.
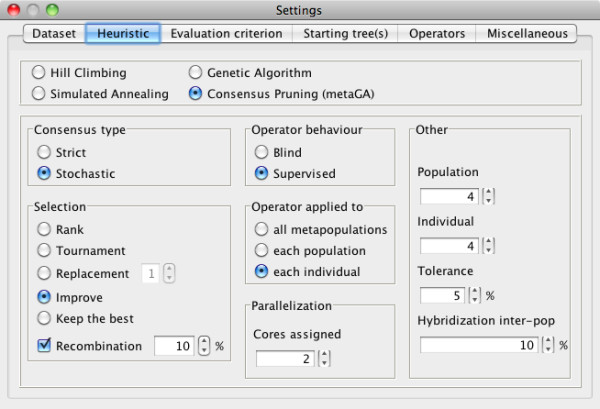
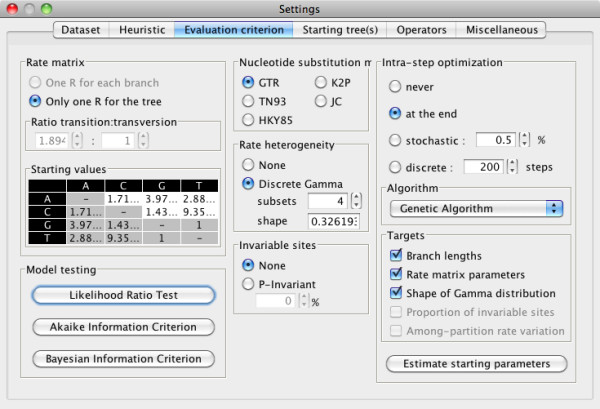
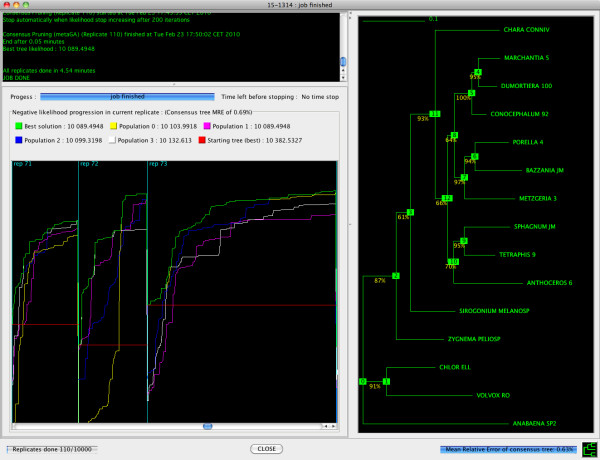
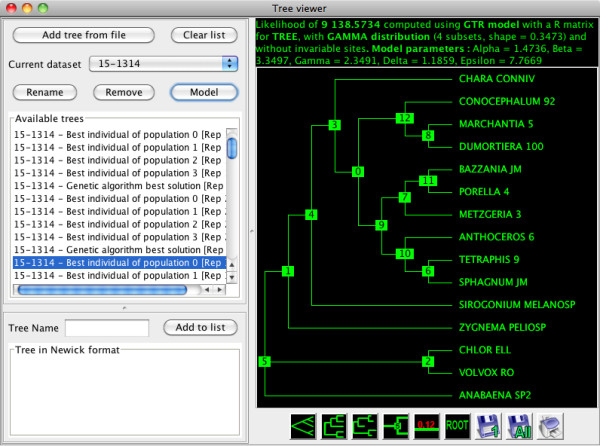
Results: Here, we present MetaPIGA v2.0, a robust implementation of several stochastic heuristics for large phylogeny inference (under maximum likelihood), including a Simulated Annealing algorithm, a classical Genetic Algorithm, and the Metapopulation Genetic Algorithm (metaGA) together with complex substitution models, discrete Gamma rate heterogeneity, and the possibility to partition data. MetaPIGA v2.0 also implements the Likelihood Ratio Test, the Akaike Information Criterion, and the Bayesian Information Criterion for automated selection of substitution models that best fit the data. Heuristics and substitution models are highly customizable through manual batch files and command line processing. However, MetaPIGA v2.0 also offers an extensive graphical user interface for parameters setting, generating and running batch files, following run progress, and manipulating result trees. MetaPIGA v2.0 uses standard formats for data sets and trees, is platform independent, runs in 32 and 64-bits systems, and takes advantage of multiprocessor and multicore computers.
Conclusions: The metaGA resolves the major problem inherent to classical Genetic Algorithms by maintaining high inter-population variation even under strong intra-population selection. Implementation of the metaGA together with additional stochastic heuristics into a single software will allow rigorous optimization of each heuristic as well as a meaningful comparison of performances among these algorithms. MetaPIGA v2.0 gives access both to high customization for the phylogeneticist, as well as to an ergonomic interface and functionalities assisting the non-specialist for sound inference of large phylogenetic trees using nucleotide sequences. MetaPIGA v2.0 and its extensive user-manual are freely available to academics at http://www.metapiga.org.
Figures

References
-
- Li W-H. Molecular evolution. Sunderland, MA.: Sinauer; 1997.
-
- Cassens I, Vicario S, Waddell VG, Balchowsky H, Van Belle D, Ding W, Fan C, Mohan RS, Simoes-Lopes PC, Bastida R. Independent adaptation to riverine habitats allowed survival of ancient cetacean lineages. Proc Natl Acad Sci USA. 2000;97(21):11343–11347. doi: 10.1073/pnas.97.21.11343. - DOI - PMC - PubMed
-
- Thorne JL, Kishino H, Painter IS. Estimating the rate of evolution of the rate of molecular evolution. Molecular Biology and Evolution. 1998;15(12):1647–1657. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
