

Familial Alzheimer's disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes
- PMID: 20634584
- PMCID: PMC4996666
- DOI: 10.3233/JAD-2010-100159
Familial Alzheimer's disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes
Abstract
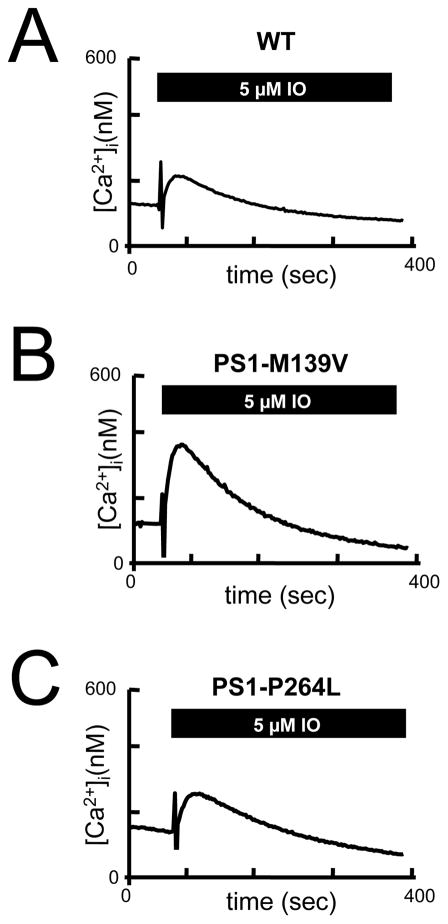
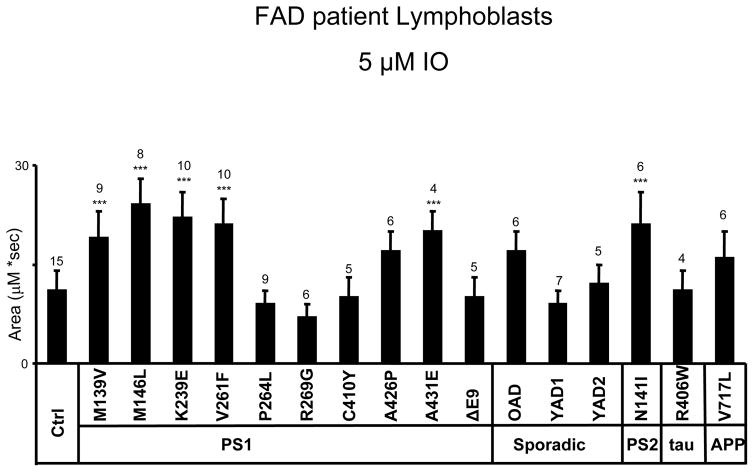
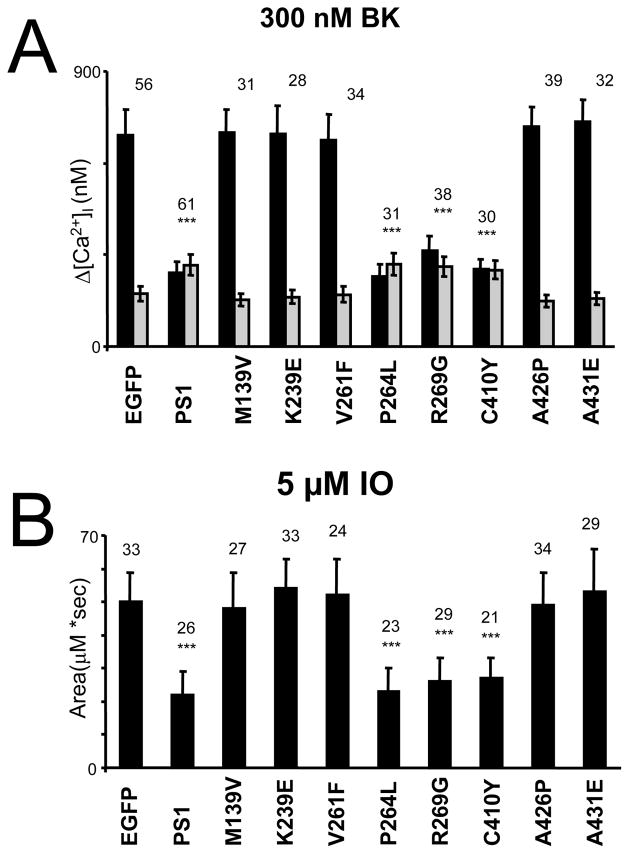
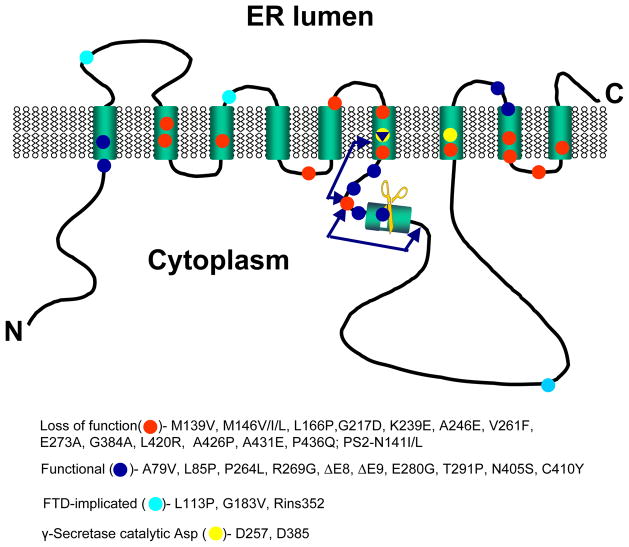
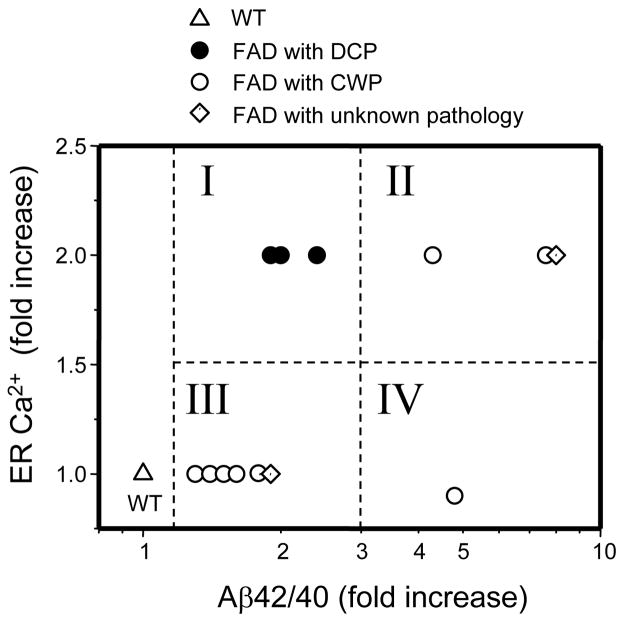
Mutations in presenilins 1 and 2 (PS1 and PS2) are responsible for approximately 40% of all early onset familial Alzheimer's disease (FAD) monogenic cases. Presenilins (PSs) function as the catalytic subunit of γ-secretase and support cleavage of the amyloid-β protein precursor (AβPP). We previously discovered that PSs also function as passive endoplasmic reticulum (ER) calcium (Ca2+) leak channels and that most FAD mutations in PSs affected their ER Ca2+ leak function. To further validate the relevance of our findings to human disease, we here performed Ca2+ imaging experiments with lymphoblasts established from FAD patients. We discovered that most FAD mutations in PSs disrupted ER Ca2+ leak function and resulted in increased ER Ca2+ pool in human lymphoblasts. However, we found that a subset of PS1 FAD mutants supported ER Ca2+ leak activity, as ER Ca2+ pool was unaffected in lymphoblasts. Most of the "functional" mutations for ER Ca2+ leak were clustered in the exon 8-9 area of PSEN1 gene and segregated with the cotton wool plaques and spastic paraparesis clinical phenotype occasionally observed in PS1 FAD patients. Our findings with the "functional" and "non-functional" PS1 FAD mutants were confirmed in Ca2+ rescue experiments with PS double-knockout mouse embryonic fibroblasts. Based on the combined effects of the PS1 FAD mutations on ER Ca2+ leak and γ-secretase activities we propose a model that explains the heterogeneity observed in FAD. The proposed model has implications for understanding the pathogenesis of both familial and sporadic AD.
Figures

References
-
- Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. - PubMed
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. - PubMed
-
- Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768–778. - PubMed
-
- Bergmans BA, De Strooper B. gamma-secretases: from cell biology to therapeutic strategies. Lancet Neurol. 2010;9:215–226. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
