

SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder
- PMID: 20637498
- PMCID: PMC2940322
- DOI: 10.1016/j.cell.2010.06.001
SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder
Abstract
N-linked glycosylation is the most frequent modification of secreted and membrane-bound proteins in eukaryotic cells, disruption of which is the basis of the congenital disorders of glycosylation (CDGs). We describe a new type of CDG caused by mutations in the steroid 5alpha-reductase type 3 (SRD5A3) gene. Patients have mental retardation and ophthalmologic and cerebellar defects. We found that SRD5A3 is necessary for the reduction of the alpha-isoprene unit of polyprenols to form dolichols, required for synthesis of dolichol-linked monosaccharides, and the oligosaccharide precursor used for N-glycosylation. The presence of residual dolichol in cells depleted for this enzyme suggests the existence of an unexpected alternative pathway for dolichol de novo biosynthesis. Our results thus suggest that SRD5A3 is likely to be the long-sought polyprenol reductase and reveal the genetic basis of one of the earliest steps in protein N-linked glycosylation.
Copyright 2010 Elsevier Inc. All rights reserved.
Figures
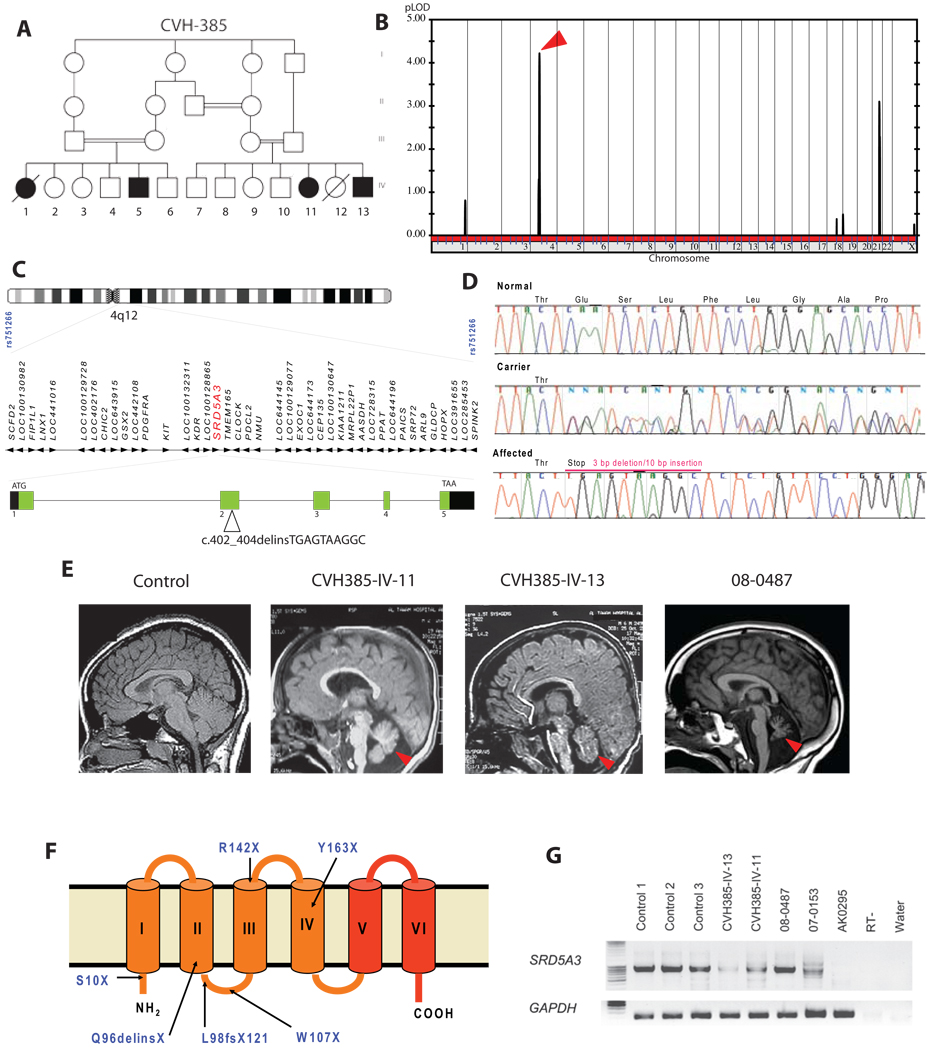
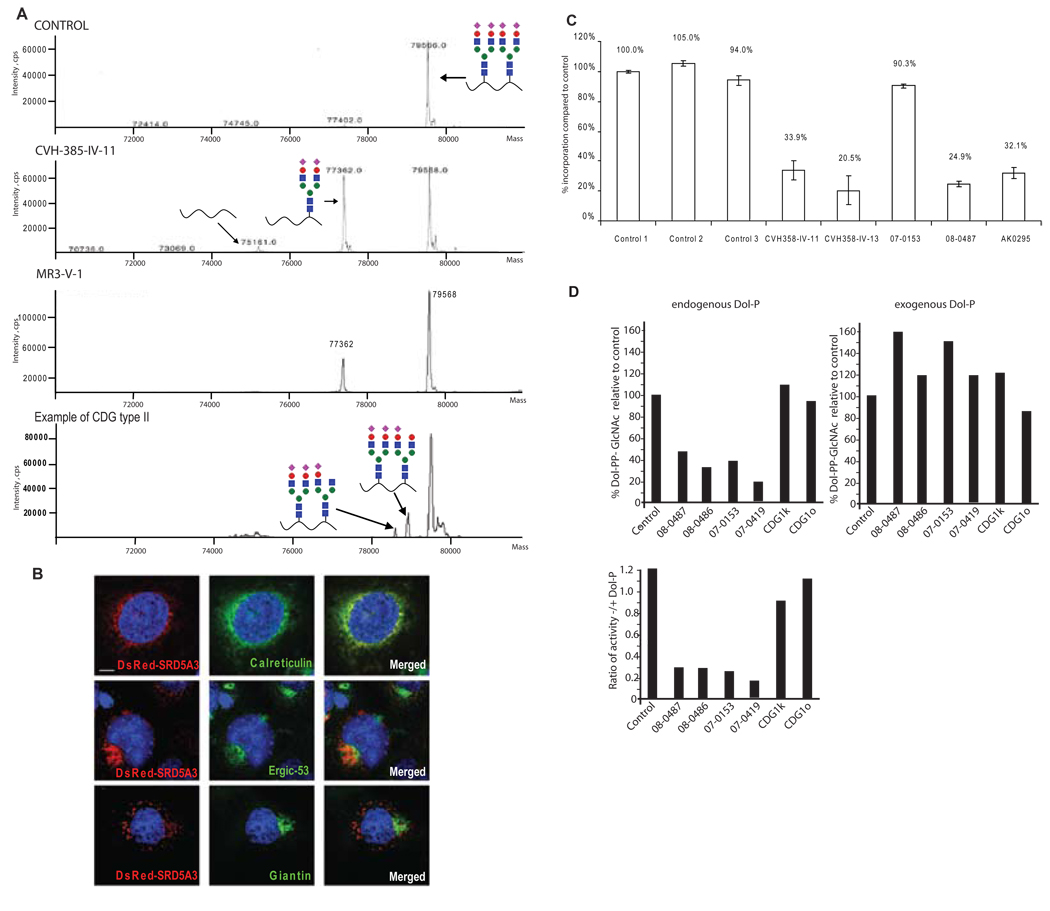
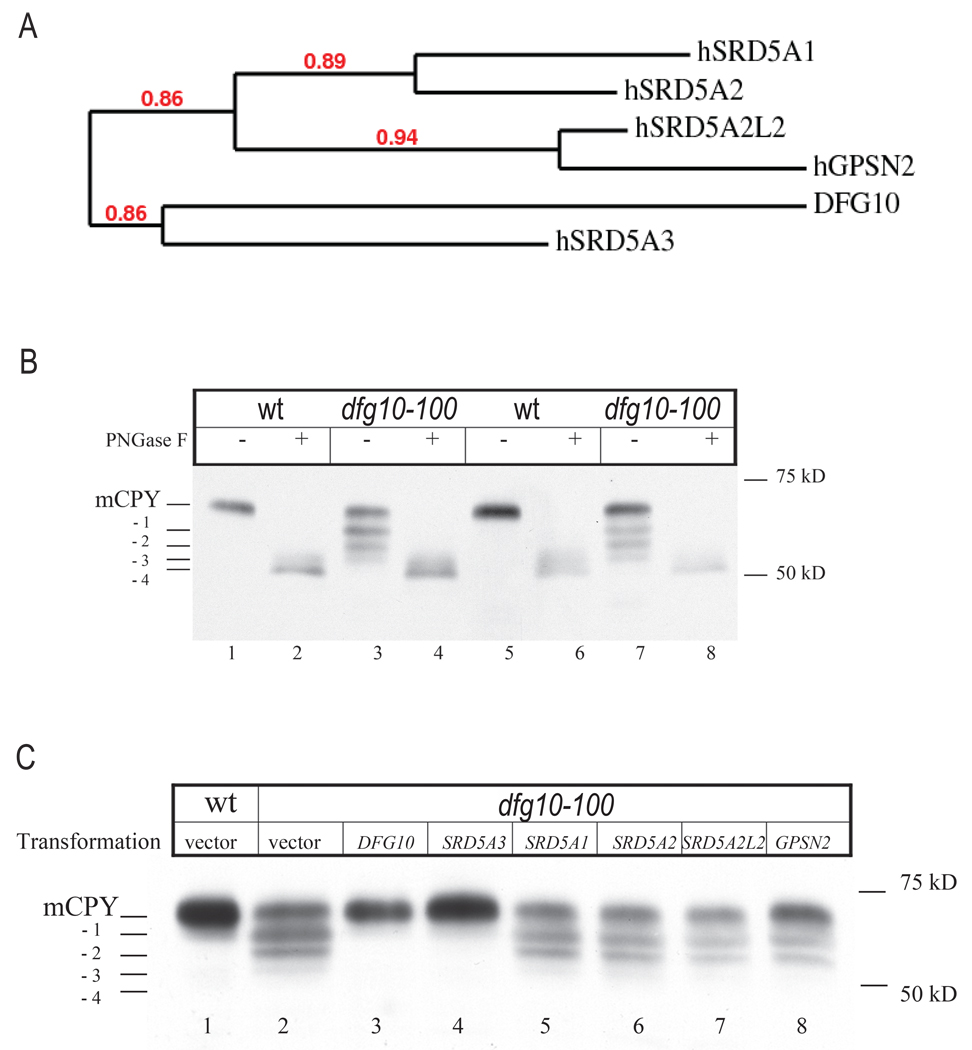
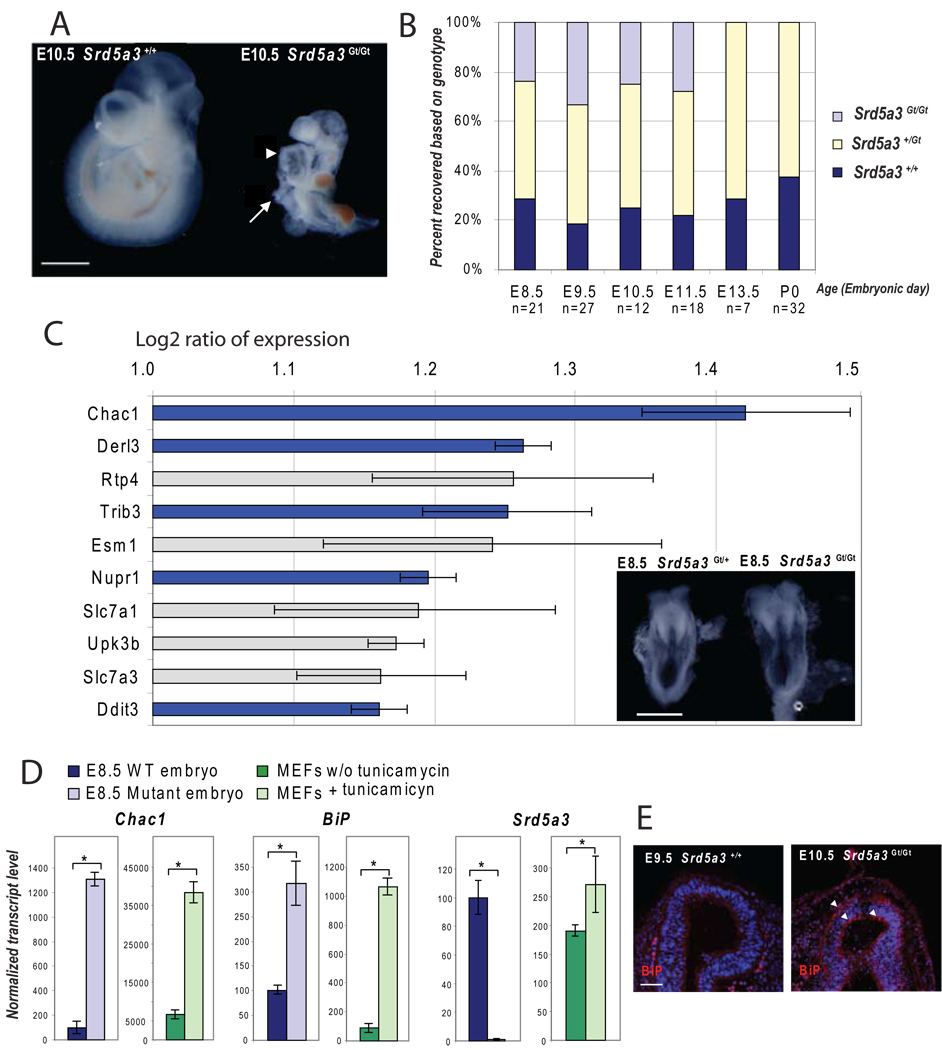
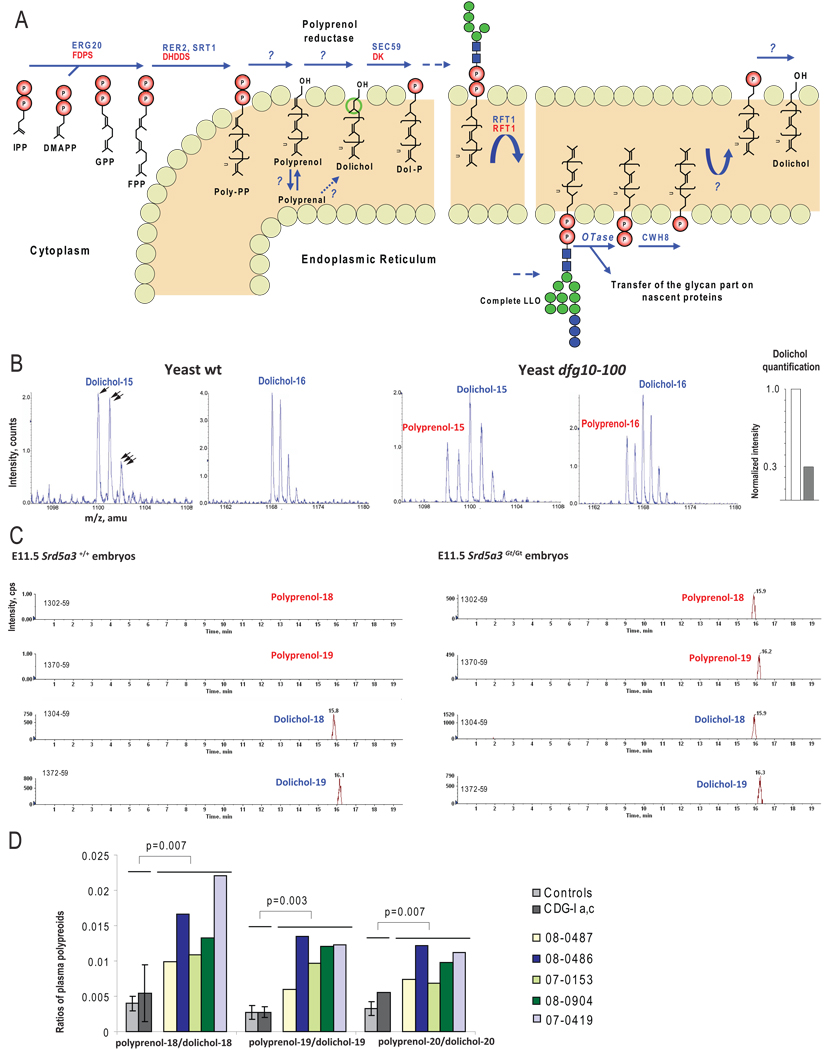
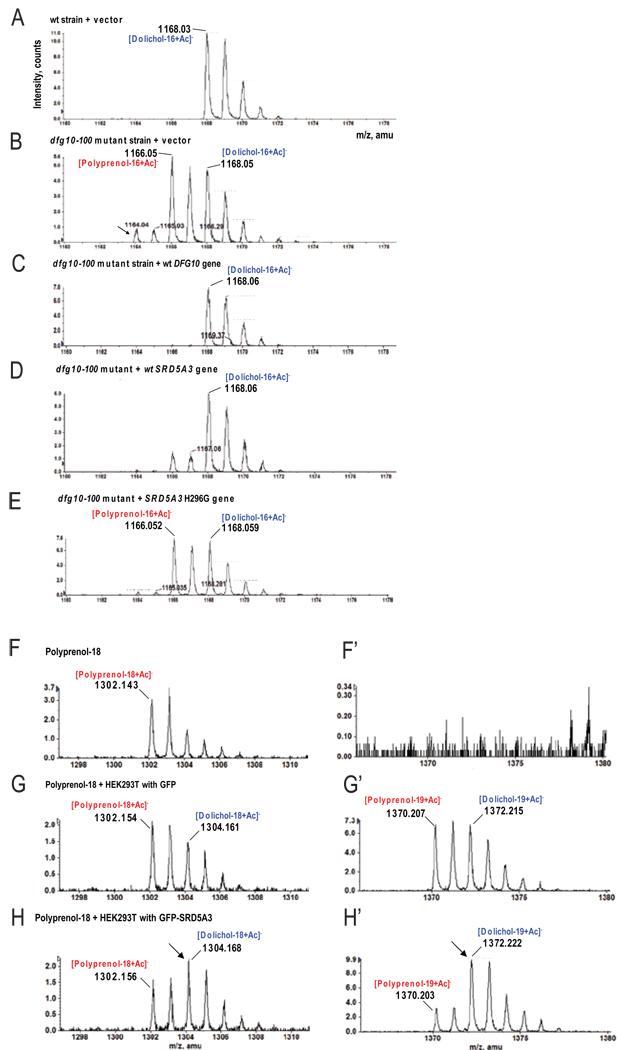
Comment in
-
SRD5A3: A surprising role in glycosylation.Cell. 2010 Jul 23;142(2):196-8. doi: 10.1016/j.cell.2010.07.003. Cell. 2010. PMID: 20655462 Free PMC article.
References
-
- Aebi M, Hennet T. Congenital disorders of glycosylation: genetic model systems lead the way. Trends Cell Biol. 2001;11:136–141. - PubMed
-
- Aggarwal S, Thareja S, Verma A, Bhardwaj TR, Kumar M. An overview on 5alpha-reductase inhibitors. Steroids. 2009 - PubMed
-
- Al-Gazali L, Hertecant J, Algawi K, El Teraifi H, Dattani M. A new autosomal recessive syndrome of ocular colobomas, ichthyosis, brain malformations and endocrine abnormalities in an inbred Emirati family. Am J Med Genet A. 2008;146:813–819. - PubMed
-
- Alber T, Kawasaki G. Nucleotide sequence of the triose phosphate isomerase gene of Saccharomyces cerevisiae. J Mol Appl Genet. 1982;1:419–434. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
