

Insulin signaling meets mitochondria in metabolism
- PMID: 20638297
- PMCID: PMC3994704
- DOI: 10.1016/j.tem.2010.06.005
Insulin signaling meets mitochondria in metabolism
Abstract
Insulin controls nutrient and metabolic homeostasis via the IRS-PI3K-AKT signaling cascade that targets FOXO1 and mTOR. Mitochondria, as the prime metabolic platform, malfunction during insulin resistance in metabolic diseases. However, the molecular link between insulin resistance and mitochondrial dysfunction remains undefined. Here we review recent studies on insulin action and the mechanistic association with mitochondrial metabolism. These studies suggest that insulin signaling underpins mitochondrial electron transport chain integrity and activity by suppressing FOXO1/HMOX1 and maintaining the NAD(+)/NADH ratio, the mediator of the SIRT1/PGC1α pathway for mitochondrial biogenesis and function. Mitochondria generate moderately reactive oxygen species (ROS) and enhance insulin sensitivity upon redox regulation of protein tyrosine phosphatase and insulin receptor. However, chronic exposure to high ROS levels could alter mitochondrial function and thereby cause insulin resistance.
Copyright © 2010. Published by Elsevier Ltd.
Figures
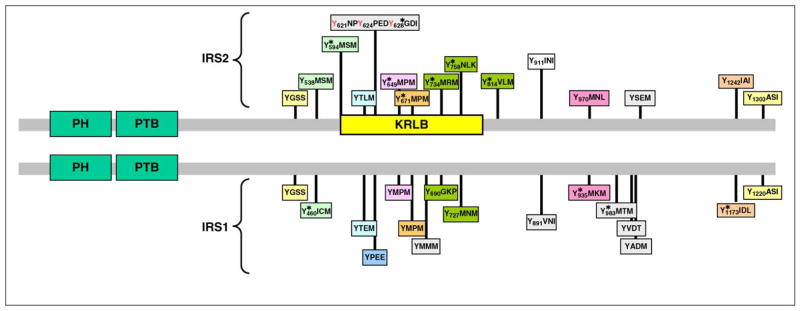
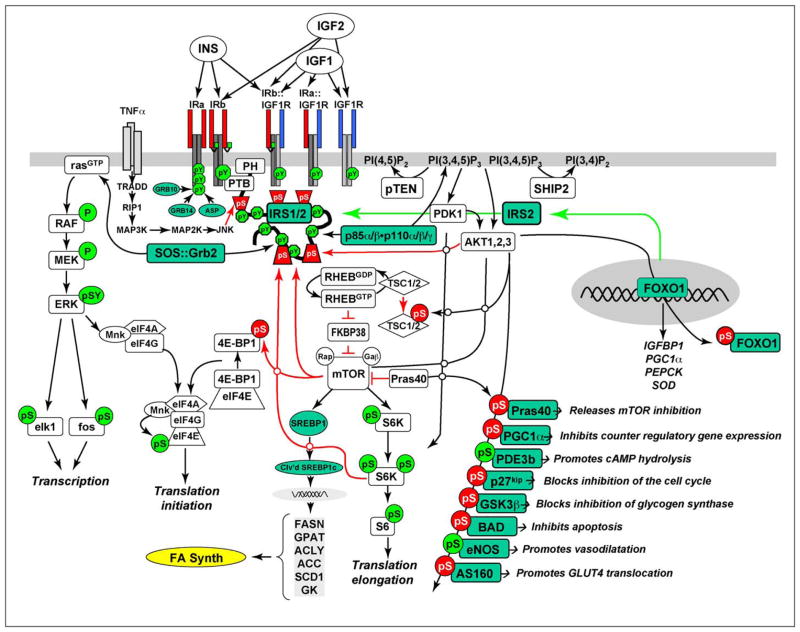
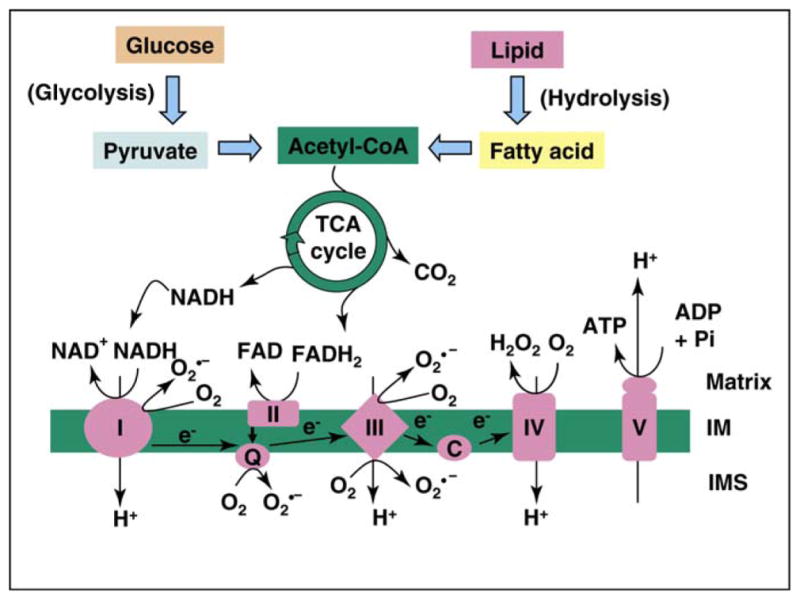
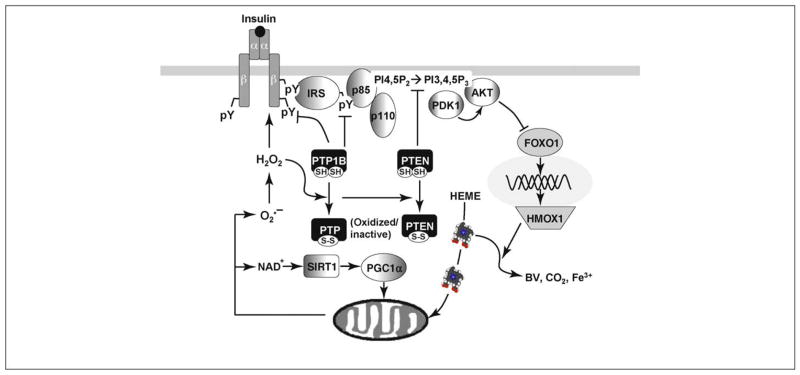
References
-
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. - PubMed
-
- Taguchi A, White MF. Insulin-like signaling, nutrient homeostasis, and life span. Annu Rev Physiol. 2008;70:191–212. - PubMed
-
- Numan S, Russell DS. Discrete expression of insulin receptor substrate-4 mRNA in adult rat brain. Brain Res Mol Brain Res. 1999;72:97–102. - PubMed
-
- Bjornholm M, et al. Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia. 2002;45:1697–1702. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
