

14-3-3gamma mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage
- PMID: 20639859
- PMCID: PMC2924644
- DOI: 10.1038/emboj.2010.157
14-3-3gamma mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage
Abstract
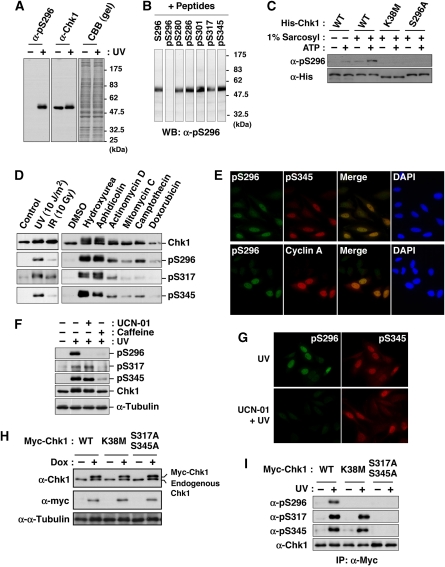
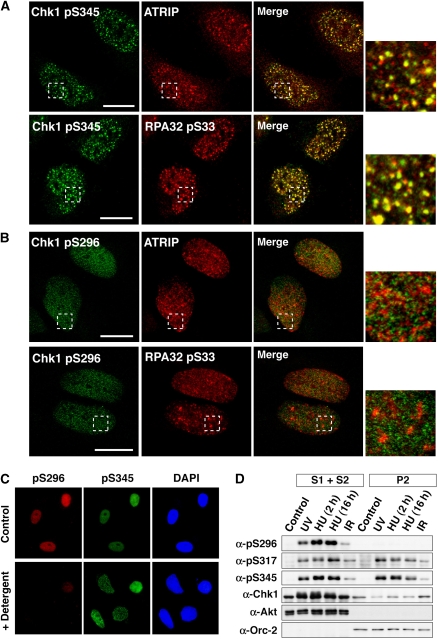
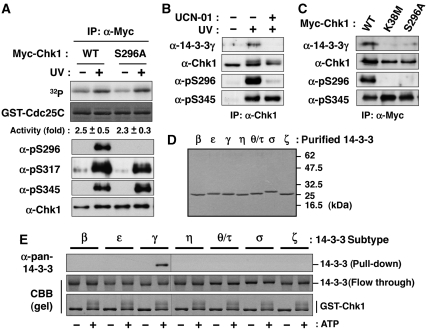
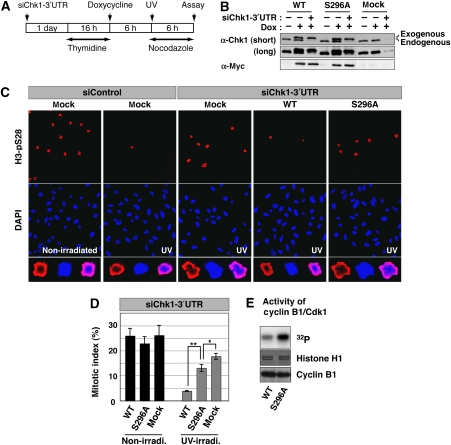
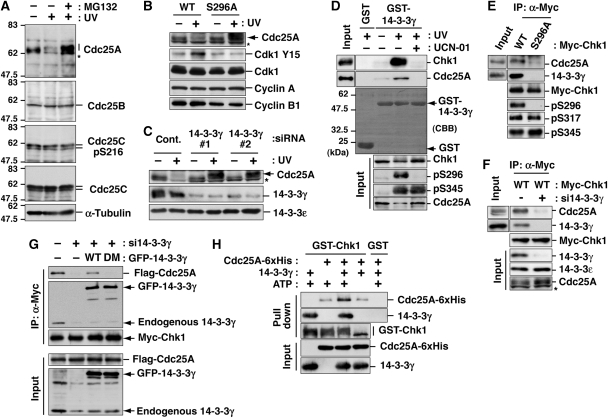
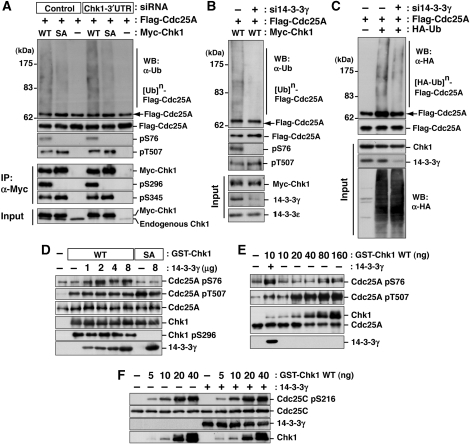
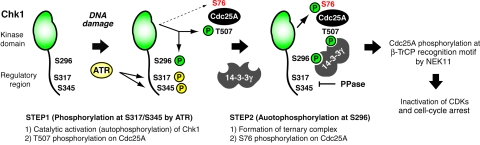
14-3-3 proteins control various cellular processes, including cell cycle progression and DNA damage checkpoint. At the DNA damage checkpoint, some subtypes of 14-3-3 (beta and zeta isoforms in mammalian cells and Rad24 in fission yeast) bind to Ser345-phosphorylated Chk1 and promote its nuclear retention. Here, we report that 14-3-3gamma forms a complex with Chk1 phosphorylated at Ser296, but not at ATR sites (Ser317 and Ser345). Ser296 phosphorylation is catalysed by Chk1 itself after Chk1 phosphorylation by ATR, and then ATR sites are rapidly dephosphorylated on Ser296-phosphorylated Chk1. Although Ser345 phosphorylation is observed at nuclear DNA damage foci, it occurs more diffusely in the nucleus. The replacement of endogenous Chk1 with Chk1 mutated at Ser296 to Ala induces premature mitotic entry after ultraviolet irradiation, suggesting the importance of Ser296 phosphorylation in the DNA damage response. Although Ser296 phosphorylation induces the only marginal change in Chk1 catalytic activity, 14-3-3gamma mediates the interaction between Chk1 and Cdc25A. This ternary complex formation has an essential function in Cdc25A phosphorylation and degradation to block premature mitotic entry after DNA damage.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 - PubMed
-
- Boutros R, Lobjois V, Ducommun B (2007) CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer 7: 495–507 - PubMed
-
- Busino L, Chiesa M, Draetta GF, Donzelli M (2004) Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene 23: 2050–2056 - PubMed
-
- Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF (2003) Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature 426: 87–91 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
