

TRAIL treatment provokes mutations in surviving cells
- PMID: 20639907
- PMCID: PMC2997681
- DOI: 10.1038/onc.2010.242
TRAIL treatment provokes mutations in surviving cells
Abstract
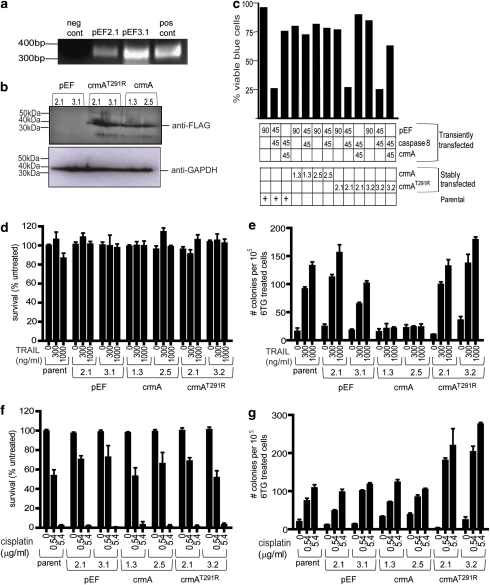
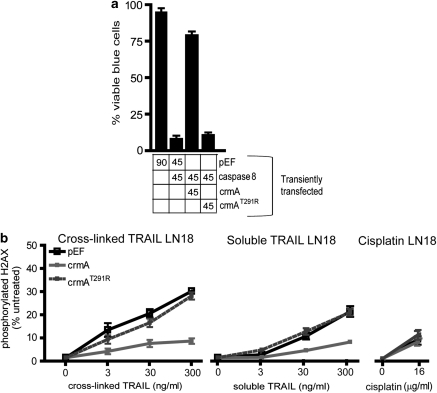
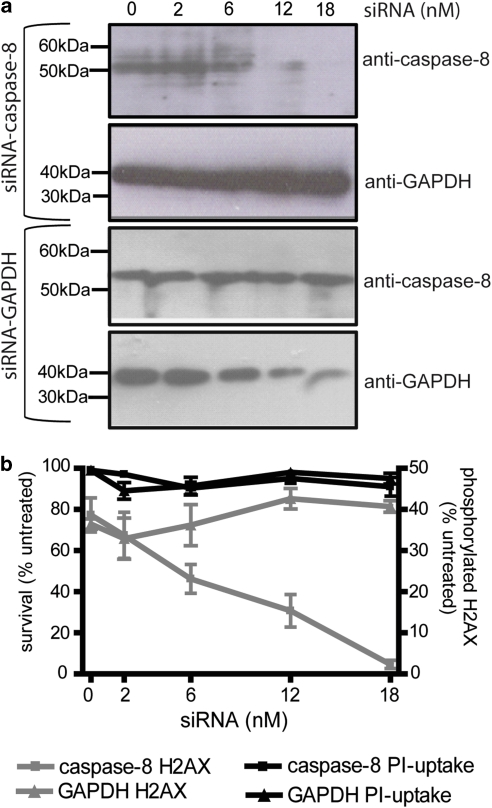
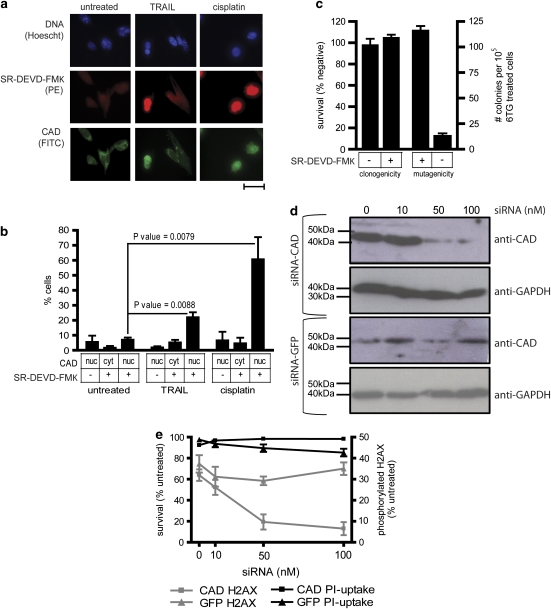
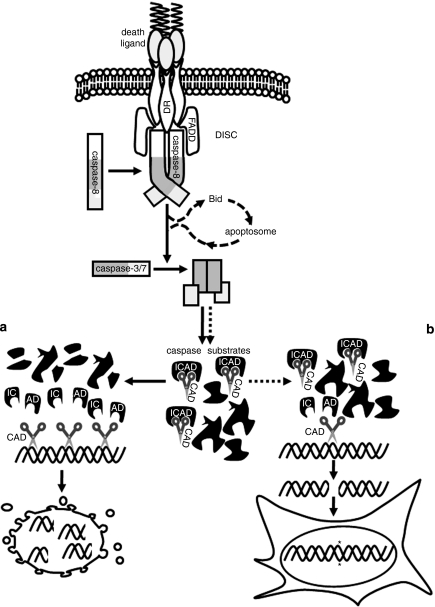
Chemotherapy and radiotherapy commonly damage DNA and trigger p53-dependent apoptosis through intrinsic apoptotic pathways. Two unfortunate consequences of this mechanism are resistance due to blockade of p53 or intrinsic apoptosis pathways, and mutagenesis of non-malignant surviving cells which can impair cellular function or provoke second malignancies. Death ligand-based drugs, such as tumor necrosis factor-related apoptosis inducing ligand (TRAIL), stimulate extrinsic apoptotic signaling, and may overcome resistance to treatments that induce intrinsic apoptosis. As death receptor ligation does not damage DNA as a primary mechanism of pro-apoptotic action, we hypothesized that surviving cells would remain genetically unscathed, suggesting that death ligand-based therapies may avoid some of the adverse effects associated with traditional cancer treatments. Surprisingly, however, treatment with sub-lethal concentrations of TRAIL or FasL was mutagenic. Mutations arose in viable cells that contained active caspases, and overexpression of the caspase-8 inhibitor crmA or silencing of caspase-8 abolished TRAIL-mediated mutagenesis. Downregulation of the apoptotic nuclease caspase-activated DNAse (CAD)/DNA fragmentation factor 40 (DFF40) prevented the DNA damage associated with TRAIL treatment. Although death ligands do not need to damage DNA in order to induce apoptosis, surviving cells nevertheless incur DNA damage after treatment with these agents.
Figures
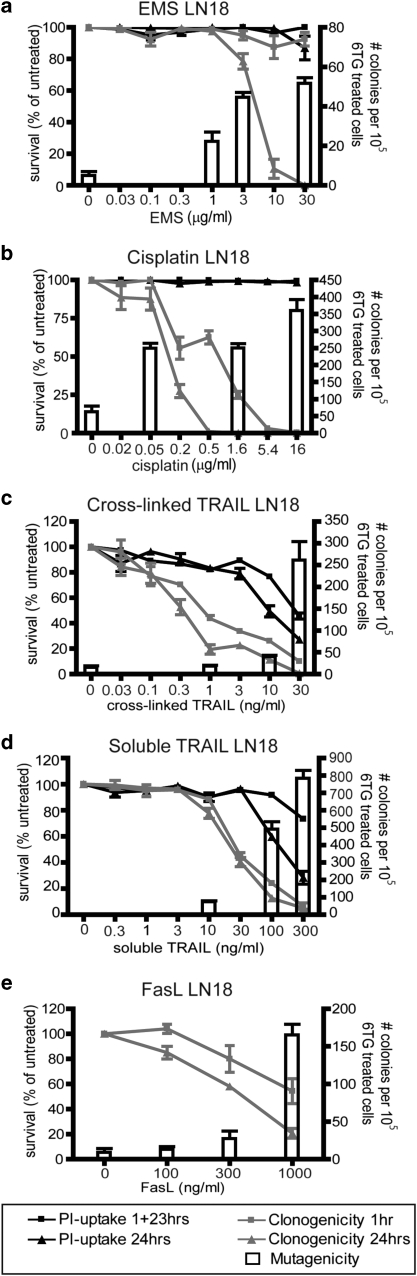
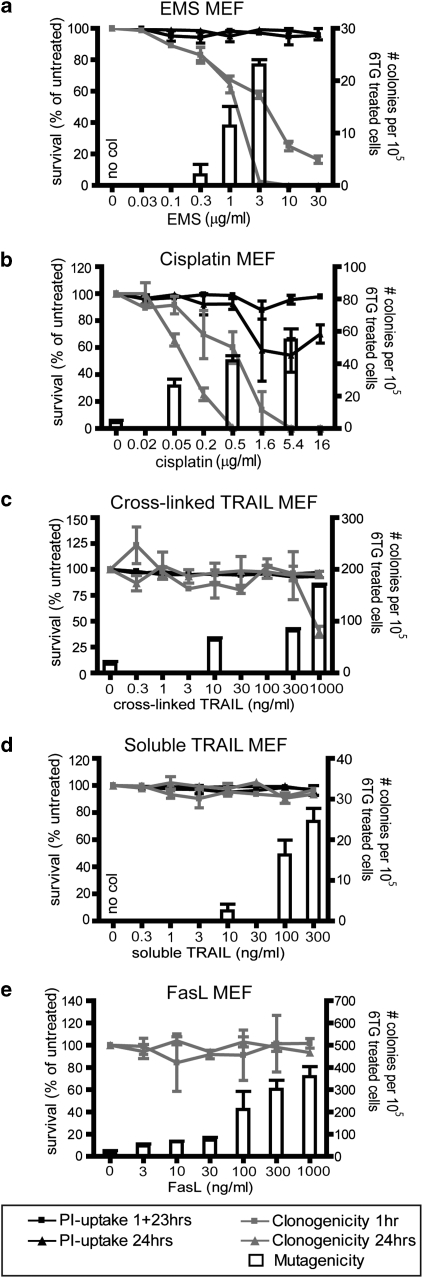
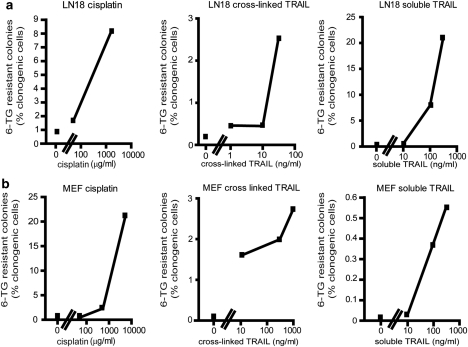
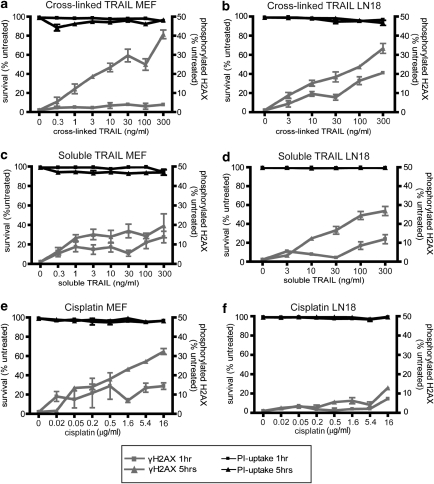
Similar articles
-
Executioner caspases and CAD are essential for mutagenesis induced by TRAIL or vincristine.Cell Death Dis. 2017 Oct 5;8(10):e3062. doi: 10.1038/cddis.2017.454. Cell Death Dis. 2017. PMID: 28981092 Free PMC article.
-
Human melanoma cells selected for resistance to apoptosis by prolonged exposure to tumor necrosis factor-related apoptosis-inducing ligand are more vulnerable to necrotic cell death induced by cisplatin.Clin Cancer Res. 2006 Feb 15;12(4):1355-64. doi: 10.1158/1078-0432.CCR-05-2084. Clin Cancer Res. 2006. PMID: 16489094
-
[Molecular mechanism of cisplatin to enhance the ability of TRAIL in reversing multidrug resistance in gastric cancer cells].Zhonghua Zhong Liu Za Zhi. 2015 Jun;37(6):404-11. Zhonghua Zhong Liu Za Zhi. 2015. PMID: 26463141 Chinese.
-
Death receptor ligands, in particular TRAIL, to overcome drug resistance.Cancer Metastasis Rev. 2001;20(1-2):51-6. doi: 10.1023/a:1013112624971. Cancer Metastasis Rev. 2001. PMID: 11831647 Review.
-
Modulation of death receptor pathways in oncology.Drugs Today (Barc). 2003;39 Suppl C:95-109. Drugs Today (Barc). 2003. PMID: 14988748 Review.
Cited by
-
Lyapunov exponents and phase diagrams reveal multi-factorial control over TRAIL-induced apoptosis.Mol Syst Biol. 2011 Nov 22;7:553. doi: 10.1038/msb.2011.85. Mol Syst Biol. 2011. PMID: 22108795 Free PMC article.
-
Mutagenic Consequences of Sublethal Cell Death Signaling.Int J Mol Sci. 2021 Jun 7;22(11):6144. doi: 10.3390/ijms22116144. Int J Mol Sci. 2021. PMID: 34200309 Free PMC article. Review.
-
Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3.Oncogene. 2011 Nov 17;30(46):4678-86. doi: 10.1038/onc.2011.185. Epub 2011 Jun 6. Oncogene. 2011. PMID: 21643018 Free PMC article.
-
Targeting cell death signalling in cancer: minimising 'Collateral damage'.Br J Cancer. 2016 Jun 28;115(1):5-11. doi: 10.1038/bjc.2016.111. Epub 2016 May 3. Br J Cancer. 2016. PMID: 27140313 Free PMC article. Review.
-
Intracellular nicotinamide adenine dinucleotide promotes TNF-induced necroptosis in a sirtuin-dependent manner.Cell Death Differ. 2016 Jan;23(1):29-40. doi: 10.1038/cdd.2015.60. Epub 2015 May 22. Cell Death Differ. 2016. PMID: 26001219 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
