

Integration and visualization of systems biology data in context of the genome
- PMID: 20642854
- PMCID: PMC2912892
- DOI: 10.1186/1471-2105-11-382
Integration and visualization of systems biology data in context of the genome
Abstract
Background: High-density tiling arrays and new sequencing technologies are generating rapidly increasing volumes of transcriptome and protein-DNA interaction data. Visualization and exploration of this data is critical to understanding the regulatory logic encoded in the genome by which the cell dynamically affects its physiology and interacts with its environment.
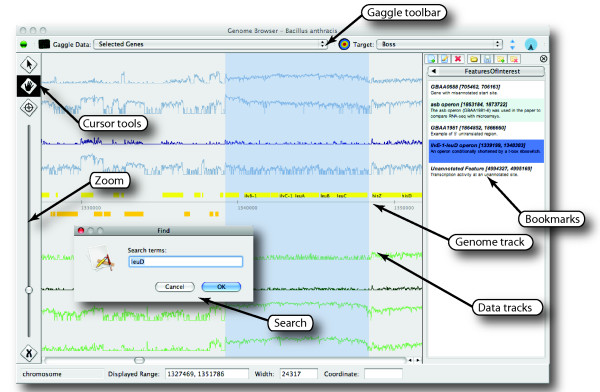
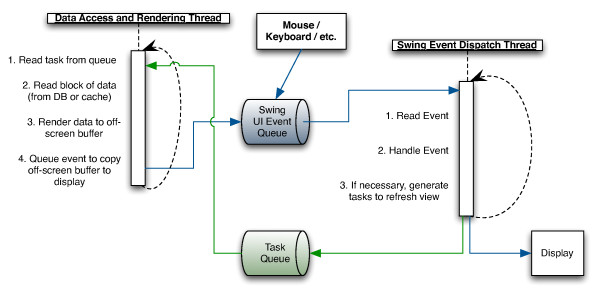
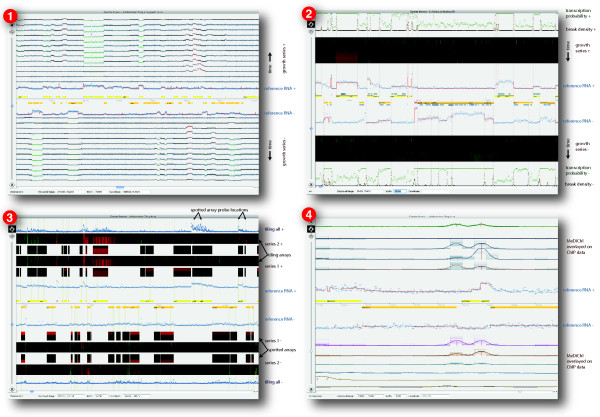
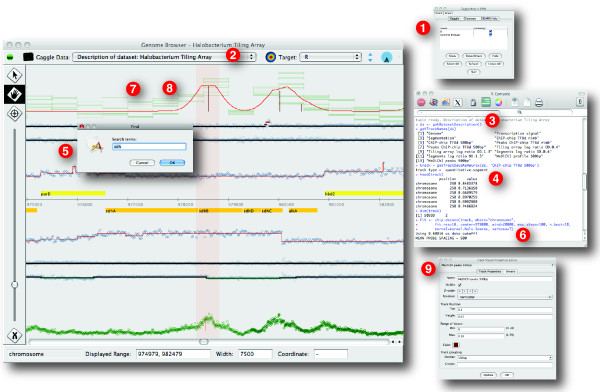
Results: The Gaggle Genome Browser is a cross-platform desktop program for interactively visualizing high-throughput data in the context of the genome. Important features include dynamic panning and zooming, keyword search and open interoperability through the Gaggle framework. Users may bookmark locations on the genome with descriptive annotations and share these bookmarks with other users. The program handles large sets of user-generated data using an in-process database and leverages the facilities of SQL and the R environment for importing and manipulating data.A key aspect of the Gaggle Genome Browser is interoperability. By connecting to the Gaggle framework, the genome browser joins a suite of interconnected bioinformatics tools for analysis and visualization with connectivity to major public repositories of sequences, interactions and pathways. To this flexible environment for exploring and combining data, the Gaggle Genome Browser adds the ability to visualize diverse types of data in relation to its coordinates on the genome.
Conclusions: Genomic coordinates function as a common key by which disparate biological data types can be related to one another. In the Gaggle Genome Browser, heterogeneous data are joined by their location on the genome to create information-rich visualizations yielding insight into genome organization, transcription and its regulation and, ultimately, a better understanding of the mechanisms that enable the cell to dynamically respond to its environment.
Figures


References
-
- Bonneau R, Facciotti MT, Reiss DJ, Schmid AK, Pan M, Kaur A, Thorsson V, Shannon P, Johnson MH, Bare JC, Longabaugh W, Vuthoori M, Whitehead K, Madar A, Suzuki L, Mori T, Chang D, Diruggiero J, Johnson CH, Hood L, Baliga NS. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. doi: 10.1016/j.cell.2007.10.053. - DOI - PubMed
-
- SQLite. http://www.sqlite.org/
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
