

Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR data
- PMID: 20643085
- PMCID: PMC2905107
- DOI: 10.1016/j.bpj.2010.04.062
Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR data
Abstract
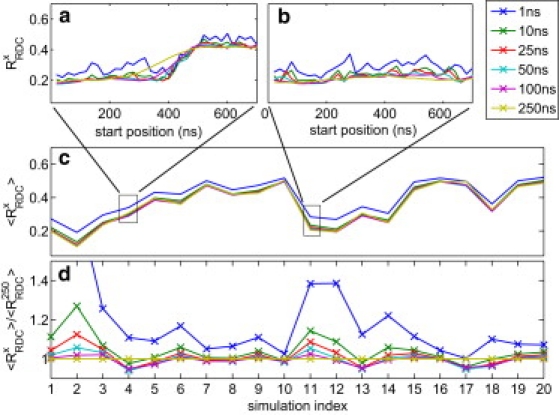
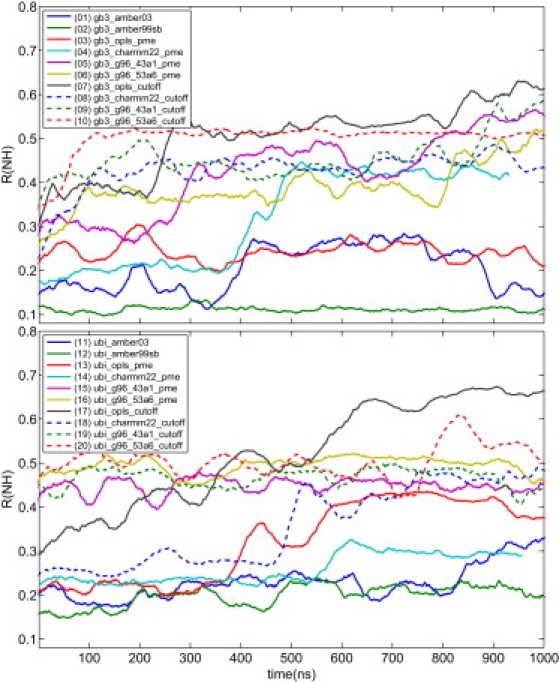
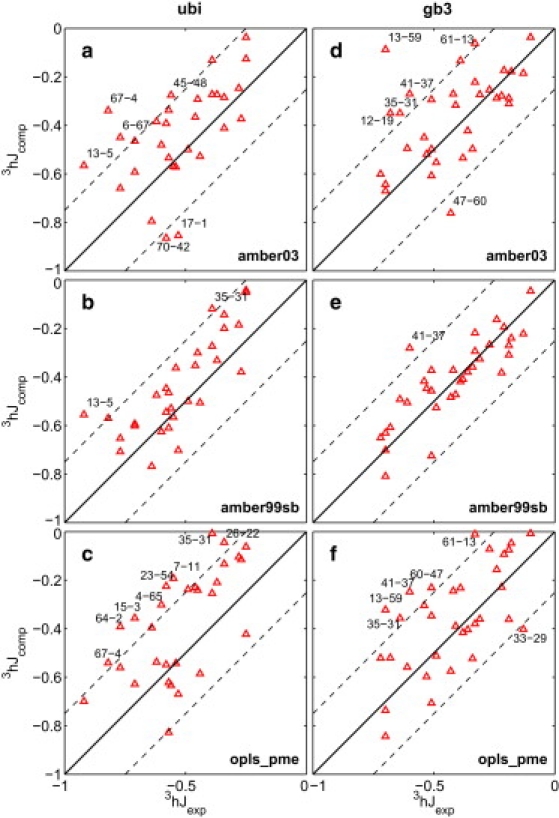
Protein dynamics on the atomic level and on the microsecond timescale has recently become accessible from both computation and experiment. To validate molecular dynamics (MD) at the submicrosecond timescale against experiment we present microsecond MD simulations in 10 different force-field configurations for two globular proteins, ubiquitin and the gb3 domain of protein G, for which extensive NMR data is available. We find that the reproduction of the measured NMR data strongly depends on the chosen force field and electrostatics treatment. Generally, particle-mesh Ewald outperforms cut-off and reaction-field approaches. A comparison to measured J-couplings across hydrogen bonds suggests that there is room for improvement in the force-field description of hydrogen bonds in most modern force fields. Our results show that with current force fields, simulations beyond hundreds of nanoseconds run an increased risk of undergoing transitions to nonnative conformational states or will persist within states of high free energy for too long, thus skewing the obtained population frequencies. Only for the AMBER99sb force field have such transitions not been observed. Thus, our results have significance for the interpretation of data obtained with long MD simulations, for the selection of force fields for MD studies and for force-field development. We hope that this comprehensive benchmark based on NMR data applied to many popular MD force fields will serve as a useful resource to the MD community. Finally, we find that for gb3, the force-field AMBER99sb reaches comparable accuracy in back-calculated residual dipolar couplings and J-couplings across hydrogen bonds to ensembles obtained by refinement against NMR data.
Copyright (c) 2010 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures



References
-
- Palmer A.G. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004;104:3623–3640. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
