

Estradiol stimulates capillary formation by human endothelial progenitor cells: role of estrogen receptor-{alpha}/{beta}, heme oxygenase 1, and tyrosine kinase
- PMID: 20644008
- PMCID: PMC2936691
- DOI: 10.1161/HYPERTENSIONAHA.110.153262
Estradiol stimulates capillary formation by human endothelial progenitor cells: role of estrogen receptor-{alpha}/{beta}, heme oxygenase 1, and tyrosine kinase
Abstract
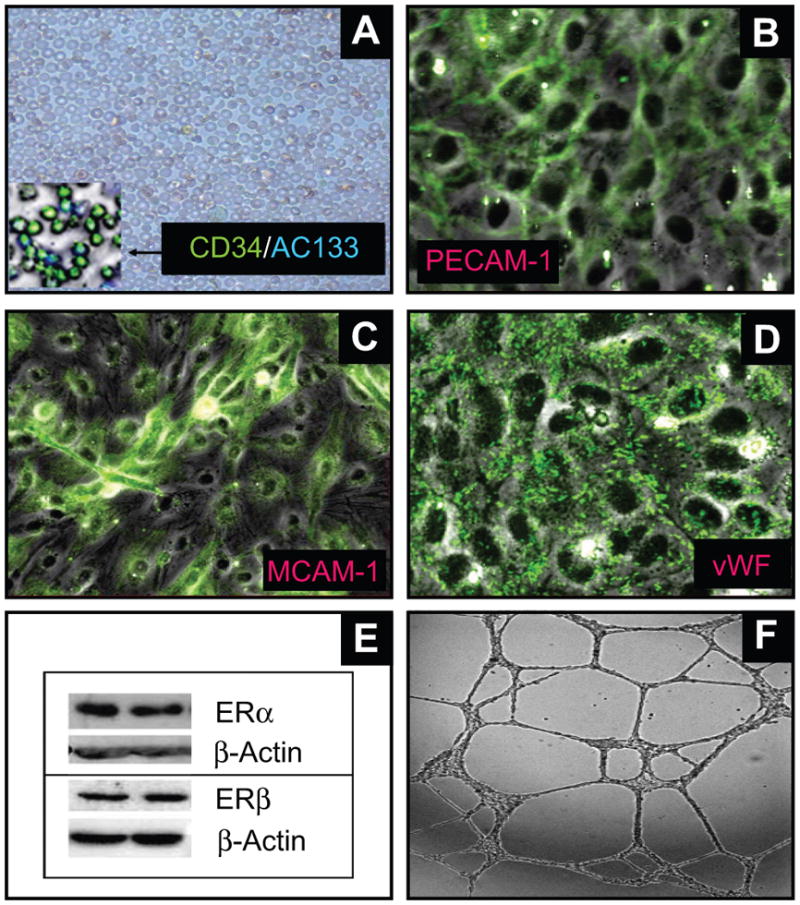
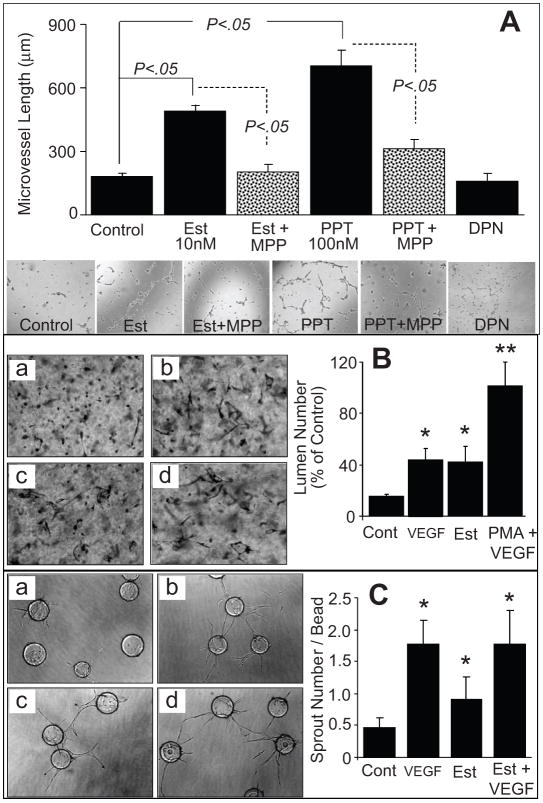
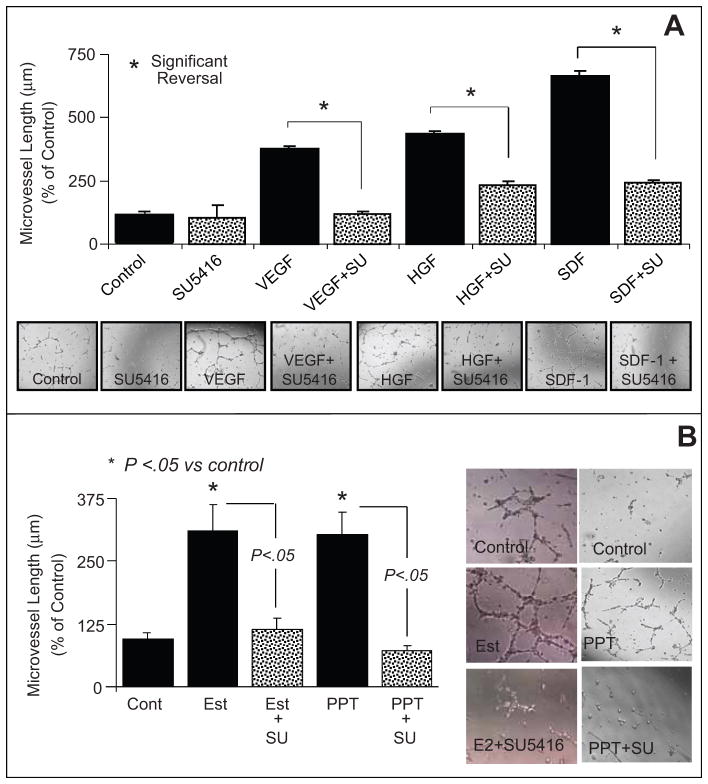
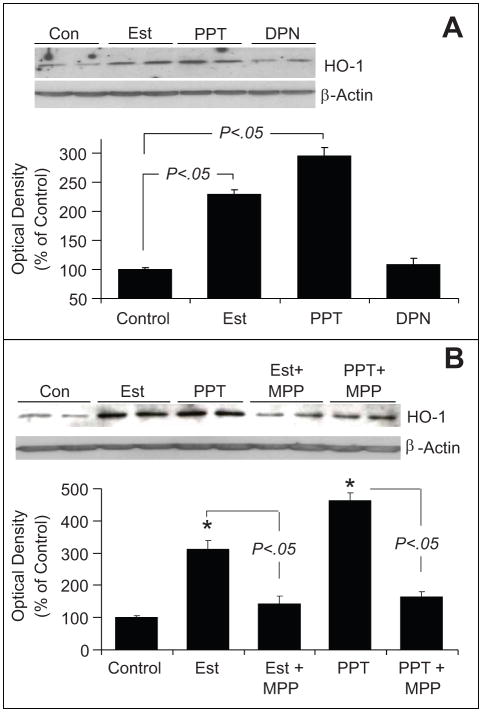
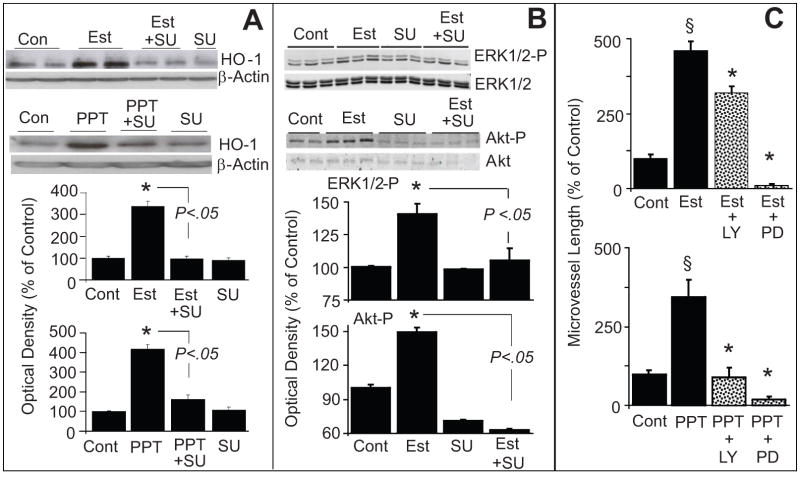
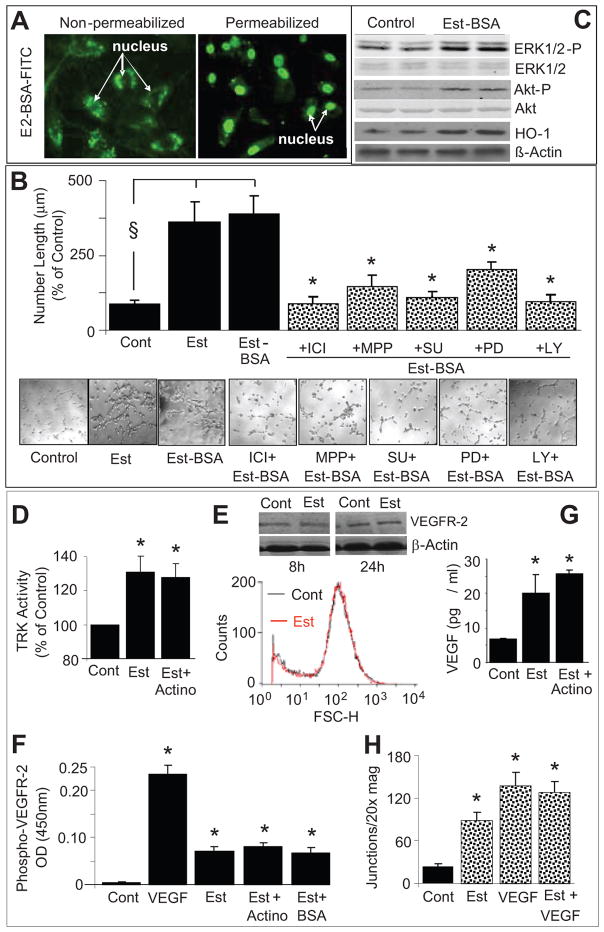
Endothelial progenitor cells (EPCs) repair damaged endothelium and promote capillary formation, processes involving receptor tyrosine kinases (RTKs) and heme oxygenase 1 (HO-1). Because estradiol augments vascular repair, we hypothesize that estradiol increases EPC proliferation and capillary formation via RTK activation and induction of HO-1. Physiological concentrations of estradiol (10 nmol/L) increased EPC-induced capillary sprout and lumen formation in matrigel/fibrin/collagen systems. Propyl-pyrazole-triol (PPT; 100 nmol/L; estrogen receptor [ER]-alpha agonist), but not diarylpropionitrile (ER-beta agonist), mimicked the stimulatory effects of estradiol on capillary formation, and methyl-piperidino-pyrazole (ER-alpha antagonist) abolished the effects of estradiol and PPT. Three different RTK activators (vascular endothelial growth factor, hepatocyte growth factor, and stromal derived growth factor 1) mimicked the capillary-stimulating effects of estradiol and PPT. SU5416 (RTK inhibitor) blocked the stimulatory effects of estradiol and PPT on capillary formation. Estradiol increased HO-1 expression by 2- to 3-fold, an effect blocked by SU5416, and PPT mimicked the effects of estradiol on HO-1. The ability of estradiol to enhance capillary formation, increase expression of HO-1, and augment phosphorylation of extracellular signal-regulated kinase 1/2, Akt, and vascular endothelial growth factor receptor 2 was mimicked by its cell-impermeable analog BSA estradiol. Actinomycin (transcription inhibitor) did not alter the effects of estradiol on RTK activity or vascular endothelial growth factor secretion. We conclude that estradiol via ER-alpha promotes EPC-mediated capillary formation by a mechanism that involves nongenomic activation of RTKs and HO-1 activation. Estradiol in particular and ER-alpha agonists in general may promote healing of injured vascular beds by promoting EPC activity leading to more rapid endothelial recovery and capillary formation after injury.
Figures
References
-
- Dubey RK, Jackson EK. Estrogen-induced cardiorenal protection: potential cellular, biochemical, and molecular mechanisms. Am J Physiol Renal Physiol. 2001;280:F365–F388. - PubMed
-
- Dubey RK, Imthurn B, Barton M, Jackson EK. Vascular consequences of menopause and hormone therapy: importance of timing of treatment and type of estrogen. Cardiovasc Res. 2005;66:295–306. - PubMed
-
- Clarkson TB, Appt SE. Coronary artery disease and postmenopausal hormone therapy: is there a time for prevention? Gynaecol Forum. 2004;9:11–14.
-
- Hodis HN, Mack WJ, Lobo RA, Shoupe D, Sevanian A, Mahrer PR, Selzer RH, Liu CrCR, Liu ChCH, Azen SP. Estrogen in the prevention of atherosclerosis. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2001;135:939–953. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
