

GSK3 signalling in neural development
- PMID: 20648061
- PMCID: PMC3533361
- DOI: 10.1038/nrn2870
GSK3 signalling in neural development
Abstract
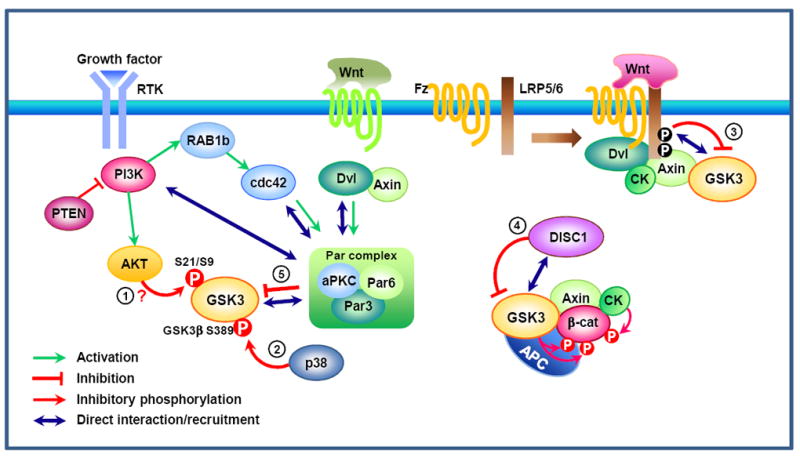
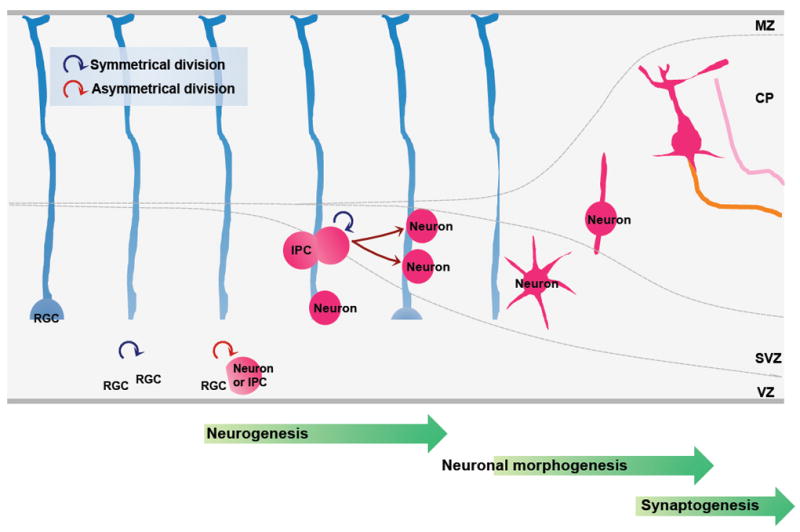
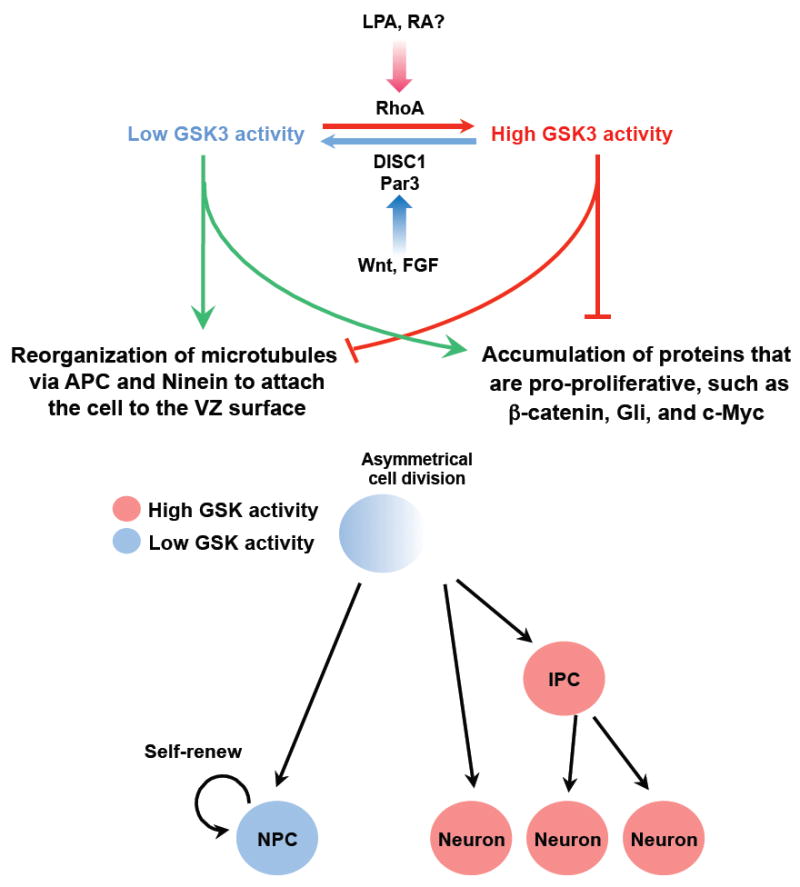
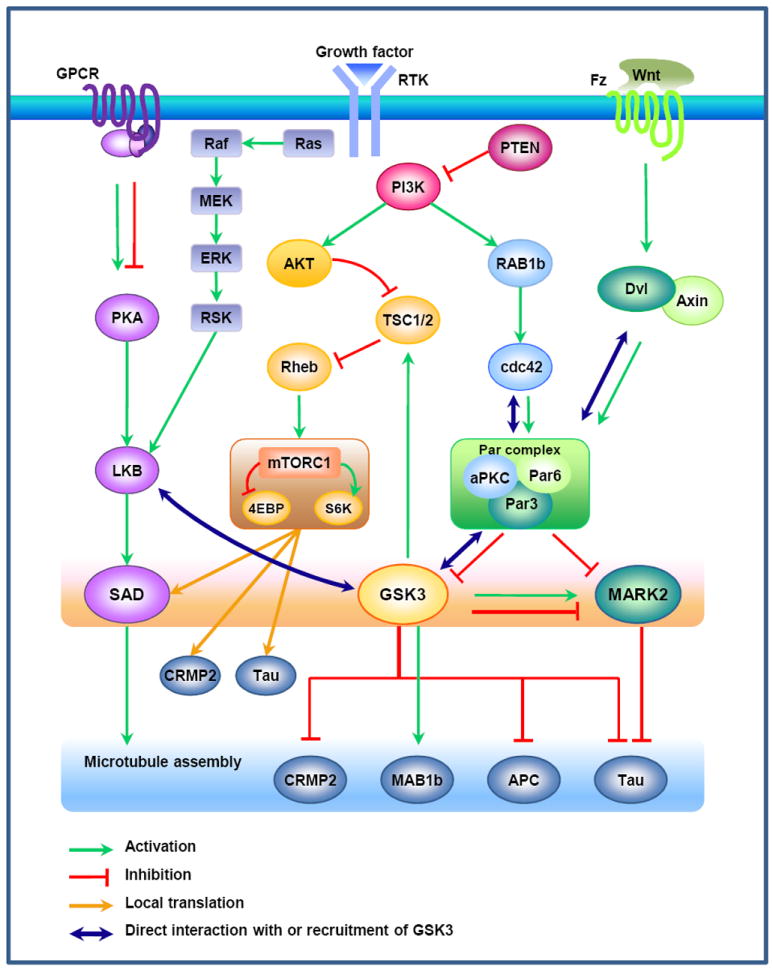
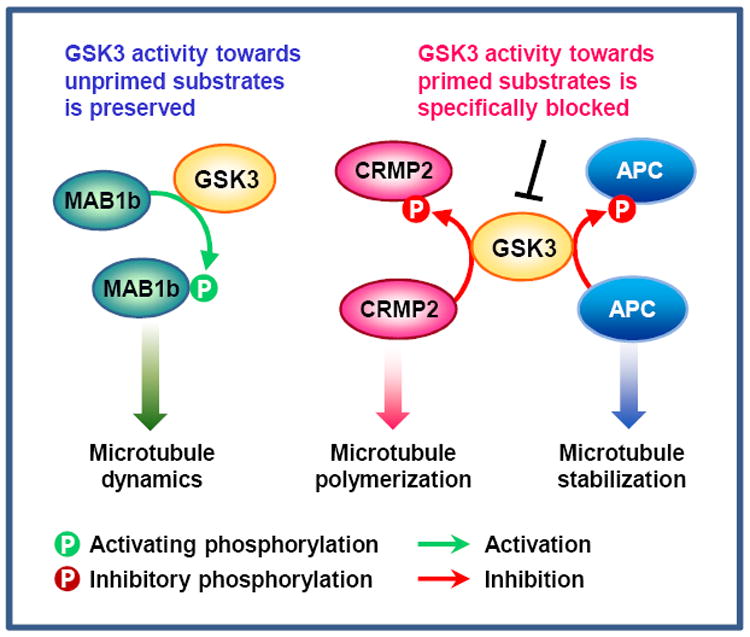
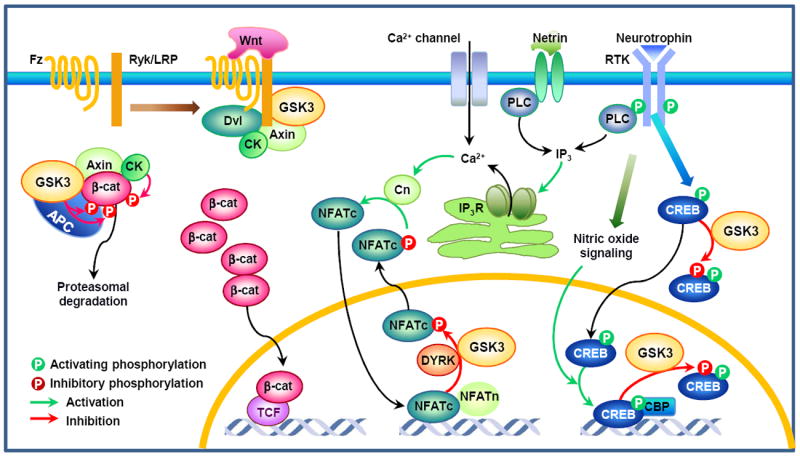
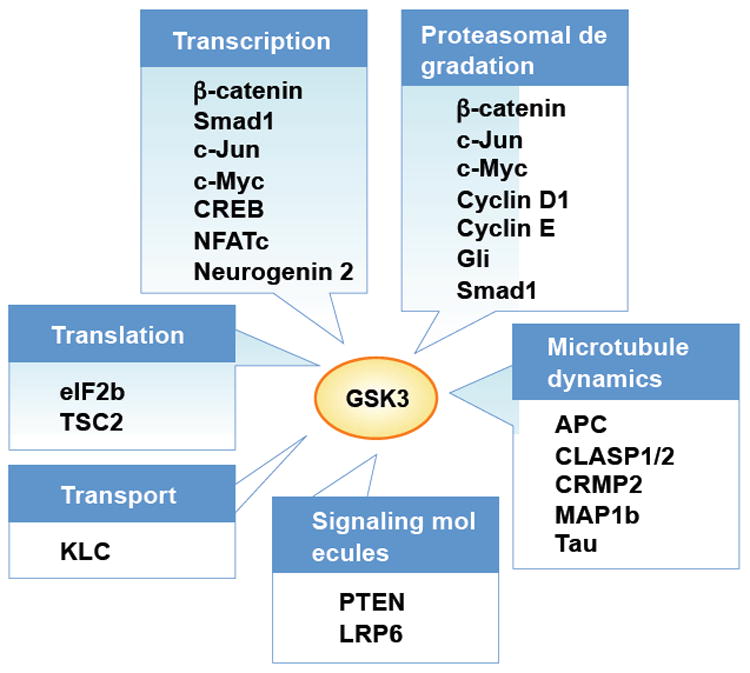
Recent evidence suggests that glycogen synthase kinase 3 (GSK3) proteins and their upstream and downstream regulators have key roles in many fundamental processes during neurodevelopment. Disruption of GSK3 signalling adversely affects brain development and is associated with several neurodevelopmental disorders. Here, we discuss the mechanisms by which GSK3 activity is regulated in the nervous system and provide an overview of the recent advances in the understanding of how GSK3 signalling controls neurogenesis, neuronal polarization and axon growth during brain development. These recent advances suggest that GSK3 is a crucial node that mediates various cellular processes that are controlled by multiple signalling molecules--for example, disrupted in schizophrenia 1 (DISC1), partitioning defective homologue 3 (PAR3), PAR6 and Wnt proteins--that regulate neurodevelopment.
Figures
References
-
- Woodgett JR, Cohen P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase-II (glycogen synthase kinase-5) Biochim Biophys Acta. 1984;788:339–47. - PubMed
-
- Wang Y, Roach PJ. Inactivation of rabbit muscle glycogen synthase by glycogen synthase kinase-3. Dominant role of the phosphorylation of Ser-640 (site-3a) J Biol Chem. 1993;268:23876–80. - PubMed
-
- Mukai F, Ishiguro K, Sano Y, Fujita SC. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J Neurochem. 2002;81:1073–83. - PubMed
-
- Castano Z, Gordon-Weeks PR, Kypta RM. The neuron-specific isoform of glycogen synthase kinase-3beta is required for axon growth. J Neurochem. 2010;113:117–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
